CRISPR Verifies Risk Alleles, Improves Gene Editing
Quick Links
New versions of the CRISPR/Cas9 gene-editing system allow researchers to alter the eukaryotic genome more precisely and efficiently than ever before. First used on the mammalian genome in 2013, CRISPR is continually being improved and is now being used to study neurodegenerative disease. In the April 20 issue of Nature, scientists led by Rudolf Jaenisch at the Whitehead Institute in Cambridge, Massachusetts, describe how they used CRISPR to introduce Parkinson’s GWAS hits into cells and measure their effect on α-synuclein expression. The paper showcases how CRISPR/Cas9 can help validate risk alleles in disease. A second paper in the same issue, by David Liu and colleagues at Harvard University, describes an improved CRISPR/Cas9 system that targets and replaces single base pairs with greater precision. This tool could in theory be used to fix pathogenic variants in the genetic code. Coupled with another new twist on CRISPR reported in the April 27 Nature, these upgrades could also simplify how scientists make animal models of disease. In that third paper, researchers led by Marc Tessier-Lavigne at Rockefeller University, New York, describe how to tweak the CRISPR system to control whether new mutations insert into the genome in homozygous or heterozygous fashion. Tessier-Lavigne will lead Stanford University as its president starting this September. These scientists introduced one or two copies of APP and presenilin FAD mutations into stem cells and found that cortical neurons derived from those cells had Aβ profiles that matched the gene dose. The method gives researchers a more accurate and efficient way to equip model systems with heterozygous mutations, which are often the cause of neurodegenerative diseases.
“The CRISPR/Cas9 system is so simple that research is progressing at an incredibly fast pace,” said Edgardo Rodriguez-Lebron, University of Florida, Gainesville. “This speaks to the beauty of the system,” he told Alzforum.

Tight Embrace.
The Cas9 protein (blue) uses CRISPR RNA (red) to target viral DNA (yellow). [Courtesy of David S. Goodsell/RCSB PDB, http://pdb101.rcsb.org/motm/motm-image-download.]
CRISPR stands for clustered regularly interspaced short palindromic repeats. In bacterial DNA, these sequences comprise genetic information left over from attacks by viruses. RNA transcribed from the repeats guides the endonuclease Cas9 in hunting down and cutting up any matching viral DNA that tries to invade again. Scientists have figured out how to co-opt this system to target Cas9 to specific sequences in the eukaryotic genome, where it slices through the double helix (see Sep 2014 series).
DNA repair mechanisms then fix the break in one of two ways. Non-homologous end-joining randomly inserts or deletes nucleotides (indels) and often causes a nonsensical frame shift, silencing the gene. Homology-directed repair (HDR), on the other hand, allows scientists to add genes. They do this by transfecting cells with a DNA strand containing 5' and 3' ends homologous to the cut site. Once Cas9 breaks the helix, the homology-directed repair system patches it with the transfected DNA, incorporating the new bit of genetic code. NHEJ is the more frequent cellular response, making CRISPR highly efficient for knocking out genes. Because HDR is much less efficient, it has been tricky to use the system to edit or add DNA. Nevertheless, CRISPR appeals to scientists because they can make guided RNA strands to match any DNA region, enabling the technology to target precise spots of the genome.
The allure of CRISPR is its potential to repair pathogenic mutations. For example, three separate groups recently used an adeno-associated virus to deliver CRISPR to the muscles of mouse models of muscular dystrophy, where it cut out mutation-carrying exons in the dystrophin gene and partially restored muscle function (Long et al., 2016; Nelson et al., 2016; Tabebordbar et al., 2016).
CRISPR Meets GWAS
Researchers have begun using the system to test the function of disease risk alleles identified in GWAS. Most GWAS hits turn up in non-coding regions of DNA, so their function is a mystery (Sep 2012 news). “There are hundreds of risk factors in Parkinson’s disease and nobody knows what they do,” Jaenisch told Alzforum. By figuring out how the risk variants influence genes, it might be possible come up with new therapeutic targets, he said.
In their paper, Jaenisch, first author Frank Soldner, and colleagues examined whether two GWAS-identified PD alleles upped expression of the α-synuclein gene (SNCA) in induced pluripotent stem cells (iPSCs). They chose to measure effects on this gene since α-synuclein inclusions are a major hallmark of the disease, and a 50 percent boost in SNCA expression is enough to trigger early onset forms of disease (Devine et al., 2011). Even subtle upticks might increase the chances of developing the late-onset form, scientists predict.
From the outset, Soldner had a problem. IPSCs—even those from cultures of the same line—can differ more than fivefold in their gene expression. How could the researchers reliably measure much smaller changes? They devised a way to compare the expression of two alleles in the same cell using different-colored primers for quantitative reverse transcription PCR. Using CRISPR, they cut the enhancer from each allele. Then they replaced one copy with either an enhancer associated with higher risk of Parkinson’s disease or one associated with lower risk. By comparing the relative rates of expression within a single cell, they sidestepped cell-to-cell variability.
Soldner and colleagues focused on alleles likely to enhance the expression of α-synuclein. They chose rs356168 in intron 4 and a microsatellite repeat region called SNCA-Rep1. They found that the risk-associated guanine at rs356168 raised α-synuclein expression by 10 percent compared with the major allele, adenosine. By contrast, the risk allele for SNCA-Rep1 had no effect on expression. Further analysis revealed that the rs356168 risk allele had weak affinity for two transcription factors that normally suppress α-synuclein expression. Without a solid hold on the gene, the repressors failed to dial down transcription as much, the researchers concluded.
“Our paper is the first one to show that a PD risk variant raises very slightly the expression of α-synuclein,” Jaenisch said. The study highlights how CRISPR/Cas9 can help scientists understand risk alleles mechanistically, moving the field toward a better grasp of sporadic disease, he said. Jaenisch plans to use this system to introduce multiple risk factors into one cell line and look at how the combined effects change the cell’s phenotype.
“This study represents a solid framework for functional follow-up on GWAS discoveries,” said Ornit Chiba-Falek, Duke University Medical Center, Durham, North Carolina. She noted that other labs are sure to conduct similar studies on genetic variants that regulate α-synuclein expression and other genes involved in neurodegenerative diseases. She suggested examining the regulation of gene expression in multiple systems, since factors such as mixed cell types and the age of a culture can influence gene expression in iPSC-derived models. “We need to interpret this result with great caution,” Chiba-Falek said. “We cannot rule out the possibility of a false negative for the SNCA-Rep1 variant finding reported here.”
A Crisper CRISPR
CRISPR-Cas9 needs improvement, particularly in controlling the indels introduced by the DNA repair systems. Is there a way to avoid the double-strand breaks that lead to indels? Liu and colleagues wrangled CRISPR to do just that. First author Alexis Komor modified Cas9 so it could only cut through one strand of the helix, then attached a deaminase that converts cytidine to uracil. When this doctored CRISPR system homes in on a particular C-G pair, it converts it to a T-A, with a little help from DNA repair machinery (see image below).
“This work represents a substantial advance in gene-editing technology; the reduction in off-target indels by avoiding double-strand breaks is a major achievement,” wrote Gary Landreth, Case Western Reserve University, Cleveland, to Alzforum. “I think this is just the beginning of where this technology is headed.”
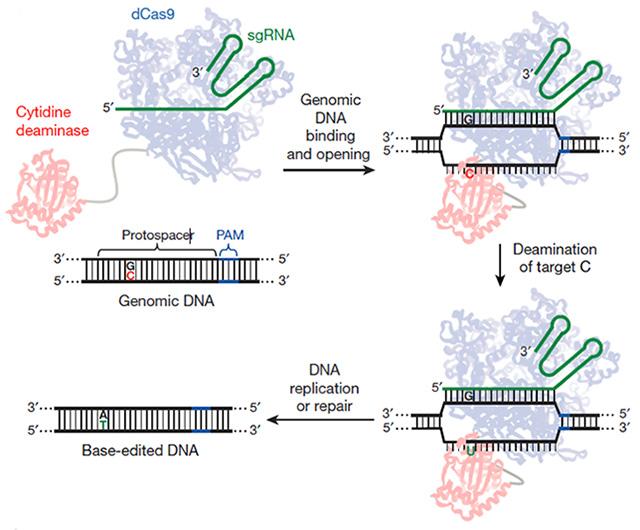
Molecular Switcheroo. This modified CRISPR/Cas9 system binds a desired spot on DNA, converts a C to a U, and lets DNA repair mechanisms replace it with a T and then pair it with an A.
The final DNA-editing machine is more efficient at replacing a single base-pair than the standard CRISPR/Cas9 system, Liu said. In HEK293T the original replaced less than 1 percent of C’s Liu targeted with T’s—the modified CRISPR replaced 37 percent. Using the new system, Komor and colleagues transformed ApoE4 to ApoE3 in mouse astrocytes by changing a C to a T in the codon for amino acid 158, changing it from arginine to cysteine. The switch occurred in up to 75 percent of cells that took up the CRISPR system. These researchers also repaired a p53 mutation in 7 percent of cells from a human breast cancer line.
Liu’s group is using this genome-editing complex to create cell and animal models of disease and is looking for enzymes or small molecules that could carry out other base conversions. Even just the C-to-T replacement could fix hundreds of disease-related genes, Liu said. “Base-editing expands the efficiency, cleanliness, and scope of gene editing for the largest class of disease-associated mutations, namely the point mutations,” Liu told Alzforum. He added that the technology is still far from clinical use, as researchers have to measure its specificity, look for off-target activity, and make sure it’s safe.
The approach significantly improves current CRISPR-Cas9 gene editing, wrote Mathew Blurton-Jones, University of California, Irvine, to Alzforum. This kind of gene editing provides a powerful tool to study specific genes, their influence on cell function, and their role in disease, he added.
A Careful CRISPR
As reported in the April 27 Nature, Tessier-Lavigne and colleagues also aimed to make CRISPR more efficient at changing genetic code. CRISPR/Cas9 is extremely active once it’s in a cell. Even if homology-directed repair incorporates a new stretch of DNA with a desired mutation into the genome, Cas9 will continuously re-cut the target site until the repair machinery snarls it with an indel, whereupon the process grinds to a halt because the guide RNA no longer matches. Co-first authors Dominik Paquet and Dylan Kwart devised a way to ensure Cas9 cuts just once. Into the DNA templates strand used for gene repair, they added a base change near the spot required to capture Cas9. After the enzyme cut the first time, HDR incorporated this DNA strand into the genome, suppressing further Cas9 cuts. This boosted HDR accuracy two- to 10-fold per allele, or up to 100-fold per cell.
In addition, the scientists wanted to control whether Cas9 modulated one or two copies of an allele in the cell. The enzyme usually modifies both. However, Paquet and colleagues capitalized on the finding that the greater the distance between Cas9’s cut and the mutation on the DNA repair template, the less likely the change was to be incorporated. By varying this “cut-to-mutation” distance, they found a sweet spot, around 10 base pairs from the double-strand break, that maximized the chances of a heterozygous change. Shorter, and a homozygous change was more likely; farther, and they might see no change at all. By designing their guide RNAs and DNA repair templates so Cas9 cut closer or farther from the desired location, the authors could cause either a homozygous or heterozygous mutation.
Paquet, Kwart, and colleagues used this approach to introduce either single or double copies of AD-related mutations to wild-type iPSCs, then derive cortical neurons. They found that homozygous APPSwe and PSEN1 M146V mutations caused a threefold rise in Aβ production or Aβ42:40 ratio, respectively. A heterozygous mutation of either one upped the value twofold. Since most patients are heterozygous for these types of mutation, these cells more closely model people’s genotypes, said Paquet. He added that these cells have advantages over those that come from patient fibroblasts. They can be developed faster, more easily differentiate into various cell types, and allow researchers to directly compare wild-type and mutant alleles in the same genetic background.
“I think these editing techniques will have the biggest impact on in vitro projects, where they will make it possible to engineer mutations in a high percentage of cells,” wrote Michael Sasner of the Jackson Laboratory, Bar Harbor, Maine, to Alzforum. They will also reduce the cost of making novel animal models with a desired point mutation. This is important when labs introduce more than one genetic change into a single model. “I’m excited to see what other base editors will become available as the CRISPR technique is developed and refined,” he wrote.—Gwyneth Dickey Zakaib
References
Series Citations
News Citations
Paper Citations
- Long C, Amoasii L, Mireault AA, McAnally JR, Li H, Sanchez-Ortiz E, Bhattacharyya S, Shelton JM, Bassel-Duby R, Olson EN. Postnatal genome editing partially restores dystrophin expression in a mouse model of muscular dystrophy. Science. 2016 Jan 22;351(6271):400-3. Epub 2015 Dec 31 PubMed.
- Nelson CE, Hakim CH, Ousterout DG, Thakore PI, Moreb EA, Castellanos Rivera RM, Madhavan S, Pan X, Ran FA, Yan WX, Asokan A, Zhang F, Duan D, Gersbach CA. In vivo genome editing improves muscle function in a mouse model of Duchenne muscular dystrophy. Science. 2016 Jan 22;351(6271):403-7. Epub 2015 Dec 31 PubMed.
- Tabebordbar M, Zhu K, Cheng JK, Chew WL, Widrick JJ, Yan WX, Maesner C, Wu EY, Xiao R, Ran FA, Cong L, Zhang F, Vandenberghe LH, Church GM, Wagers AJ. In vivo gene editing in dystrophic mouse muscle and muscle stem cells. Science. 2016 Jan 22;351(6271):407-11. Epub 2015 Dec 31 PubMed.
- Devine MJ, Gwinn K, Singleton A, Hardy J. Parkinson's disease and α-synuclein expression. Mov Disord. 2011 Oct;26(12):2160-8. PubMed.
Further Reading
Papers
- Chu VT, Weber T, Wefers B, Wurst W, Sander S, Rajewsky K, Kühn R. Increasing the efficiency of homology-directed repair for CRISPR-Cas9-induced precise gene editing in mammalian cells. Nat Biotechnol. 2015 May;33(5):543-8. Epub 2015 Mar 24 PubMed.
- Tagliafierro L, Chiba-Falek O. Up-regulation of SNCA gene expression: implications to synucleinopathies. Neurogenetics. 2016 Jul;17(3):145-57. Epub 2016 Mar 7 PubMed.
- Smith AJ, Humphries SE, Talmud PJ. Identifying functional noncoding variants from genome-wide association studies for cardiovascular disease and related traits. Curr Opin Lipidol. 2015 Apr;26(2):120-6. PubMed.
- Paul DS, Soranzo N, Beck S. Functional interpretation of non-coding sequence variation: concepts and challenges. Bioessays. 2014 Feb;36(2):191-9. Epub 2013 Dec 5 PubMed.
Primary Papers
- Soldner F, Stelzer Y, Shivalila CS, Abraham BJ, Latourelle JC, Barrasa MI, Goldmann J, Myers RH, Young RA, Jaenisch R. Parkinson-associated risk variant in distal enhancer of α-synuclein modulates target gene expression. Nature. 2016 May 5;533(7601):95-9. Epub 2016 Apr 20 PubMed.
- Komor AC, Kim YB, Packer MS, Zuris JA, Liu DR. Programmable editing of a target base in genomic DNA without double-stranded DNA cleavage. Nature. 2016 Apr 20; PubMed.
- Paquet D, Kwart D, Chen A, Sproul A, Jacob S, Teo S, Olsen KM, Gregg A, Noggle S, Tessier-Lavigne M. Efficient introduction of specific homozygous and heterozygous mutations using CRISPR/Cas9. Nature. 2016 May 5;533(7601):125-9. Epub 2016 Apr 27 PubMed.
Annotate
To make an annotation you must Login or Register.
Comments
No Available Comments
Make a Comment
To make a comment you must login or register.