Could Calming Overactive Ryanodine Receptor Restore Autophagy?
Quick Links
Overzealous calcium signaling and dysfunctional protein degradation occur early in Alzheimer’s disease pathogenesis. Now, new evidence links the two. Two recent papers support the hypothesis that, when Ca2+ leaks from the endoplasmic reticulum, it disrupts autophagy, allowing proteins, such as Aβ and tau, to accumulate (Dec 2014 conference news). Both studies identify the ryanodine receptor (RyR), a calcium release channel, as the source of the ions.
- Ca2+ released from ryanodine receptors slows autophagy.
- Amyloidosis mice have overreactive RyRs and sluggish lysosomes.
- Genetically weakening RyRs revs up autophagy, limits plaques.
- So do small molecules that calm overactive RyRs.
In the February 22 Journal of Neuroscience, scientists led by Ilya Bezprozvanny at the University of Texas Southwestern Medical Center in Dallas reported on a mouse model of amyloidosis carrying a faulty RyR. In them, as calcium flux from the ER to the cytosol slows, autophagy revs up, fewer amyloid plaques form, and synaptic plasticity is better than in mice expressing the normal RyR.
In PNAS last November 28, scientists led by Grace (Beth) Stutzmann at the Rosalind Franklin University of Medicine and Science, North Chicago, Illinois, described the molecular basis for this suppression of autophagy. They found that, in amyloidosis mice and in induced human neurons from people with AD, calcium released by RyR limits levels of the lysosomal proton pump vacuolar ATPase. The pH of lysosomes then creeps up, spoiling proteolysis and autophagy. By normalizing RyRs pharmacologically in mice, the authors restored lysosomal protease function and lowered levels of phospho-tau and Aβ. Alzforum covered some of this work at the Alzheimer’s Association International Conference in 2019 (Sep 2019 conference news).
“These papers link endoplasmic reticulum and lysosome activity, showing communication between two organelles that are critical for the homeostasis of neurons,” Wim Annaert at KU Leuven, Belgium, told Alzforum. Frédéric Checler of the French National Centre for Scientific Research in Valbonne, France, considers the findings important. “It is a huge step toward better understanding the phenotypic alterations linking calcium signaling perturbation and autophagic processes,” he wrote (comment below).
To study RyRs in amyloidosis, first author Hua Zhang of Bezprozvanny’s lab used mice carrying the E4872Q variant in RyR2, a subtype enriched in the human brain and heart. Co-author Wayne Chen, University of Calgary, Canada, engineered these animals to shorten the channel’s open time, stunting calcium ion flow from the ER to the cytosol (Chen et al., 2014). Zhang crossed these “EQ” mice with APP NL-F knock-ins or APP/PS1 transgenic mice. The knock-ins develop amyloid plaques at 6 months of age, the transgenics already at 6 weeks.
Compared to cultured hippocampal neurons from wild-type mice, those from APP knock-ins contained more cytosolic calcium and more of the autophagosome marker LC3, indicating a backup in the autophagy system. In neurons from the EQ and EQ:APPKI animals, cytosolic calcium levels were lower and autophagy appeared to be humming along normally.
Zhang saw a similar pattern in hippocampal slices from 6-month-old mice. EQ, EQ:APPKI, and EQ:APP/PS1 animals turned over LC3 faster than did wild-type RyR mice. More of the autophagy marker co-localized with LAMP1-positive lysosomes, indicating formation of healthy autophagosomes and active autophagy.
Were the two pathways linked? Indeed, autophagy ramped up in wild-type and APP knock-in cultured neurons treated with high doses of ryanodine to inhibit RyR. Ditto when the cells were given an inhibitor of calcineurin (CaN), a calcium-dependent phosphatase activated by high levels of cytosolic calcium ions. Neither EQ nor EQ;APPKI neurons responded to inhibition of either enzyme, since their RyR activity was already low. These results suggest that calcium ions released from RyR activate CaN. This, in turn, suppresses autophagy by dephosphorylating and deactivating AMP kinase, which then fails to phosphorylate ULK1, a component of the autophagy complex (see figure below). Tamping down RyR activity restores this signaling, as measured by western blot of mouse brain lysates.
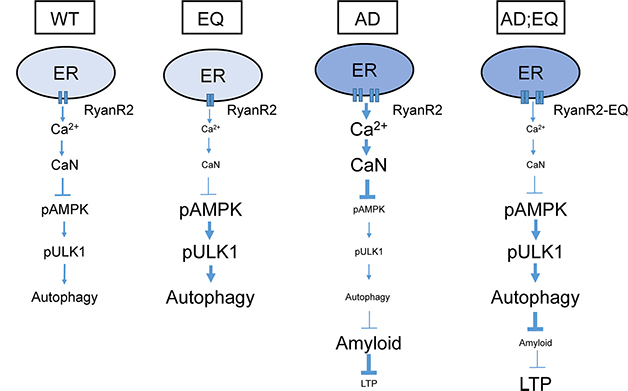
From Reticulum to Autophagy. In wild-type neurons (left) the ryanodine receptor releases calcium, stirring calcineurin to slow autophagy via pAMPK/pULK1. The E4872Q variant (EQ) reins in calcium release, lifting the calcineurin brake on pAMPK/pULK1 activation, and spurring autophagy. In AD mice, overactive RyR2 does the opposite, enabling amyloid accumulation and suppressing synaptic plasticity. The EQ mutation reverses both effects (right). [Courtesy of Zhang et al., Journal of Neuroscience, 2023.]
With autophagy ramped-up, EQ;APP/PS1 mice had fewer amyloid plaques than APP/PS1 mice (see image below). Because APP knock-ins have scant plaques at this age, the EQ RyR mutation made no difference to their amyloid load. On the other side of the coin, Chen’s group had previously reported that EQ/5xFAD crosses had just as many plaques as 5xFAD controls (Yao et al., 2020). Bezprozvanny thinks the plaque load in the 5xFAD model might be too high to be rescued by the weakened RyR.
Zhang found that synaptic plasticity, as measured by long-term potentiation in hippocampal slices, was normal in both EQ-expressing amyloidosis mice. Likewise, Chen has previously reported that hyperexcitable neurons calmed, synaptic plasticity improved, and memory was restored by expressing the EQ RyR in 5xFAD and 3xTg mice (Sun et al., 2021; Liu et al., 2021).

The EQualizer? The E4872Q RyR variant curtailed plaques in APP/PS1 mice (right) and normalized synaptic plasticity (not shown). [Courtesy of Zhang et al., Journal of Neuroscience, 2023.]
For her part, Stutzmann and colleagues dissected molecular links between RyR activity and lysosomal/autophagy dysfunction. First author Sarah Mustaly-Kalimi used 3xTg mice, which develop amyloid plaques by 6 months and, like other amyloidosis models, have overactive RyR2 channels. To see what happened when the receptor’s activity was normalized, she injected the negative allosteric modulator dantrolene into 3- to 4-month-old transgenic or wild-type mice daily for four weeks, then analyzed the brain tissue.
Compared to wild-type, the 3xTg mouse brain contained more cytosolic calcium and more LC3, i.e., sluggish autophagosomes. In contrast, dantrolene-treated 3xTG mice had wild-type calcium levels, normal autophagy, and even less S262 phosphorylated tau in their neurons. P-tau262 is an early sign of tau hyperphosphorylation in mice.
Because 3xTg mice carry a mutant version of presenilin-1, which disrupts trafficking of vacuolar ATPase subunits from the ER to lysosomes, the scientists measured the V0a1 subunit in the brain slices (Jun 2010 news). The transgenic mice had less V0a1 in the hippocampus than did wild-types, and less of it co-localized with lysosomes, which were less acidic than usual. Dantrolene restored all three readouts.
Is RyR overactive in some forms of Alzheimer's disease, then? To find out, Mustaly-Kalimi grew neurons from human induced pluripotent stem cells from nine people who had familial AD caused by the A246E presenilin-1 mutation. Indeed, these neurons churned out five times more calcium via RyRs than did neurons created from healthy adults.
As in mice, the A246E neurons had less V0a1 than did wild-type cells, and their lysosomes were less acidic. They also dampened autophagy, aggregated tau, and had an elevated Aβ42/40 ratio. Dantrolene normalized all these deficits and restored lysosomal protease activity.
All told, Stutzmann and colleagues believe that calcium released from the ER via RyR limits vATPase activity in lysosome membranes and, therefore, their acidity and proteolytic function (see image below).
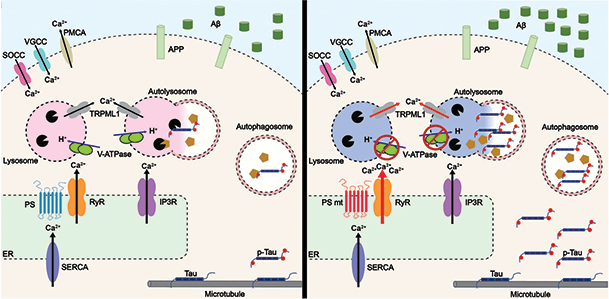
Roaring RyR. In healthy neurons (left) with wild-type presenilin (blue), ryanodine receptors (orange) release little calcium from the to the cytosol, vATPase (green ovals) acidifies lysosomes (pink circles), which degrade Aβ and phospho-tau. In AD neurons (right) expressing mutant presenilin-1 (red), overactive RyRs flood the cytosol with Ca2+, limiting vATPase trafficking to, and acidification of, lysosomes (blue circles). This allows Aβ, p-tau to accumulate. [Courtesy of Mustaly-Kalimi et al., PNAS, 2022.]
How does RyR get to be so hyper in the first place? That is unclear. Annaert agrees with both sets of authors that calcium dysregulation precedes lysosome dysfunction; however, rather than vATPase being at the root of the lysosome’s problem, he suspects that calcium released from the ER triggers faulty calcium signaling, not a pH increase, in lysosomes, (Jul 2012 news).
Ralph Nixon of New York University, Orangeburg, has a different take. He places lysosome/autophagy dysfunction upstream of RyR activity. Nixon believes that, in presenilin 1 knockout neurons, lysosomes with dysfunctional acidification wedge open TRPML1 calcium channels, causing calcium to flood the cytosol (May 2022 news; Lee et al., 2015). “Unless there are [genetic or molecular] explanations [for] the basis for the RyR activation that exclude TRPML1 triggered by pH-dysregulated lysosomes, I suspect that the data are the expected outcomes of primary lysosomal dysfunction,” Nixon wrote.
Different still is the explanation suggested by Cora O’Neill at University College Cork, Ireland, though hers also involves TRPML1. In a paper in press in the Journal of Cell Science, O'Neill reported that human induced neurons carrying APOE4, the strongest genetic risk factor for late-onset AD, release less lysosomal calcium via TRPML1 than do ApoE3, E2, and ApoE knockout neurons (Somogyi et al., 2023).
ApoE4 causes neuronal endolysosomes to swell (Nuriel et al., 2017). O'Neill suspects that sluggish TRPML1 might spur RyR to compensate by churning out more calcium into the cytosol. Such cross-talk occurs in muscle cells, though whether it happens in neurons is unknown (Thakore et al., 2020; Griffin et al., 2020; Li and Li, 2021). She thinks that targeting TRPML1 with an agonist might restore the lysosome channel activity, ease RyR overactivation—and potentially treat AD.
How about modulating RyR? “Targeting RyR needs to be handled delicately because RyR is involved in so many pathways,” cautioned Stutzmann. “You don’t want to block this channel. You want to restore its normal structure and function with a negative allosteric modulator, such as dantrolene.” Dantrolene, aka Ryanodex, was FDA approved in 2014 to treat malignant hyperthermia, a severe reaction to certain anesthetic drugs, and is being tested in some other diseases.
Dantrolene specifically binds the RyR1 subtype, which is enriched in skeletal muscle. However, when RyR2 is hyperphosphorylated, as it is in AD, its secondary structure changes, exposing a normally hidden pocket to which dantrolene binds. “This may be why we see such profound effects of dantrolene in AD but almost never in wild-type or non-AD conditions,” Stutzmann explained.—Chelsea Weidman Burke
References
News Citations
- Calcium Disruptions Wreak Synaptic Havoc
- Can Induced Neurons Identify Early Signs of Neurodegeneration?
- Death of the Neatnik: Neurons Perish When Trash Clutters Their Space?
- Presenilins and Calcium: A Lysosomal Stew With Acid Controversy
- Presenilin Mutations Stall Endosomal Transport, Swell Axons
Research Models Citations
Mutations Citations
Paper Citations
- Chen W, Wang R, Chen B, Zhong X, Kong H, Bai Y, Zhou Q, Xie C, Zhang J, Guo A, Tian X, Jones PP, O'Mara ML, Liu Y, Mi T, Zhang L, Bolstad J, Semeniuk L, Cheng H, Zhang J, Chen J, Tieleman DP, Gillis AM, Duff HJ, Fill M, Song LS, Chen SR. The ryanodine receptor store-sensing gate controls Ca2+ waves and Ca2+-triggered arrhythmias. Nat Med. 2014 Feb;20(2):184-92. Epub 2014 Jan 19 PubMed.
- Yao J, Sun B, Institoris A, Zhan X, Guo W, Song Z, Liu Y, Hiess F, Boyce AK, Ni M, Wang R, Ter Keurs H, Back TG, Fill M, Thompson RJ, Turner RW, Gordon GR, Chen SR. Limiting RyR2 Open Time Prevents Alzheimer's Disease-Related Neuronal Hyperactivity and Memory Loss but Not β-Amyloid Accumulation. Cell Rep. 2020 Sep 22;32(12):108169. PubMed.
- Sun B, Yao J, Chen AW, Estillore JP, Wang R, Back TG, Chen SR. Genetically and pharmacologically limiting RyR2 open time prevents neuronal hyperactivity of hippocampal CA1 neurons in brain slices of 5xFAD mice. Neurosci Lett. 2021 Jul 27;758:136011. Epub 2021 Jun 4 PubMed.
- Liu Y, Yao J, Song Z, Guo W, Sun B, Wei J, Estillore JP, Back TG, Chen SR. Limiting RyR2 open time prevents Alzheimer's disease-related deficits in the 3xTG-AD mouse model. J Neurosci Res. 2021 Nov;99(11):2906-2921. Epub 2021 Aug 5 PubMed.
- Lee JH, McBrayer MK, Wolfe DM, Haslett LJ, Kumar A, Sato Y, Lie PP, Mohan P, Coffey EE, Kompella U, Mitchell CH, Lloyd-Evans E, Nixon RA. Presenilin 1 Maintains Lysosomal Ca(2+) Homeostasis via TRPML1 by Regulating vATPase-Mediated Lysosome Acidification. Cell Rep. 2015 Sep 1;12(9):1430-44. Epub 2015 Aug 20 PubMed.
- Somogyi A, Kirkham ED, Lloyd-Evans E, Winston J, Allen ND, Mackrill JJ, Anderson KE, Hawkins PT, Gardiner SE, Waller-Evans H, Sims R, Boland B, O'Neill C. The synthetic TRPML1 agonist ML-SA1 rescues Alzheimer-related alterations of the endosomal-autophagic-lysosomal system. J Cell Sci. 2023 Mar 15;136(6) Epub 2023 Mar 21 PubMed.
- Nuriel T, Peng KY, Ashok A, Dillman AA, Figueroa HY, Apuzzo J, Ambat J, Levy E, Cookson MR, Mathews PM, Duff KE. The Endosomal-Lysosomal Pathway Is Dysregulated by APOE4 Expression in Vivo. Front Neurosci. 2017;11:702. Epub 2017 Dec 12 PubMed.
- Thakore P, Pritchard HA, Griffin CS, Yamasaki E, Drumm BT, Lane C, Sanders KM, Feng Earley Y, Earley S. TRPML1 channels initiate Ca2+ sparks in vascular smooth muscle cells. Sci Signal. 2020 Jun 23;13(637) PubMed.
- Griffin CS, Alvarado MG, Yamasaki E, Drumm BT, Krishnan V, Ali S, Nagle EM, Sanders KM, Earley S. The intracellular Ca2+ release channel TRPML1 regulates lower urinary tract smooth muscle contractility. Proc Natl Acad Sci U S A. 2020 Dec 1;117(48):30775-30786. Epub 2020 Nov 16 PubMed.
- Li G, Li PL. Lysosomal TRPML1 Channel: Implications in Cardiovascular and Kidney Diseases. Adv Exp Med Biol. 2021;1349:275-301. PubMed.
External Citations
Further Reading
Papers
- Oulès B, Del Prete D, Greco B, Zhang X, Lauritzen I, Sevalle J, Moreno S, Paterlini-Bréchot P, Trebak M, Checler F, Benfenati F, Chami M. Ryanodine receptor blockade reduces amyloid-β load and memory impairments in Tg2576 mouse model of Alzheimer disease. J Neurosci. 2012 Aug 22;32(34):11820-34. PubMed.
Primary Papers
- Zhang H, Knight C, Chen SR, Bezprozvanny I. A Gating Mutation in Ryanodine Receptor Type 2 Rescues Phenotypes of Alzheimer's Disease Mouse Models by Upregulating Neuronal Autophagy. J Neurosci. 2023 Feb 22;43(8):1441-1454. Epub 2023 Jan 10 PubMed.
- Mustaly-Kalimi S, Gallegos W, Marr RA, Gilman-Sachs A, Peterson DA, Sekler I, Stutzmann GE. Protein mishandling and impaired lysosomal proteolysis generated through calcium dysregulation in Alzheimer's disease. Proc Natl Acad Sci U S A. 2022 Dec 6;119(49):e2211999119. Epub 2022 Nov 28 PubMed.
Annotate
To make an annotation you must Login or Register.
Comments
Institut de Pharmacologie Moléculaire et Cellulaire
Besides canonical anatomical lesions linked to Aβ peptides and hyperphosphorylated tau protein, Alzheimer’s disease is also characterized by several other dysfunctions, including in Ca2+ signaling and mitophagic/autophagic function. Although it is well documented that autophagy could be modulated by intracellular calcium, the mechanistic link remained unclear. In these two papers, the teams of Ilya Bezprozvanny and Grace Stutzmann identified the Ryanodine receptor (RyR) as the center of gravity in an interplay between intracellular calcium deficits and lysosomal/autophagic function.
The two articles nicely used distinct complementary state-of-the-art approaches. Bezprozvanny and colleagues showed that basal hippocampal RyR2 activity inhibited autophagy by a calcineurin- and AMPK-dependent cascade. They used knock-in mice, in which an activity-deficient RyR was expressed, that they crossed with an AD model displaying exacerbated amyloid-related stigmata. Interestingly, reduction of RyR rescued Aβ accumulation and abrogated synaptic plasticity defects.
In Stutzmann et al., by means of fluorescent biosensors and optical imaging, they similarly conclude on the involvement of the RyR. In neurons derived from AD patients, it associated with a reduction of the lysosomal proton pump vacuolar ATPase expression and, thereby, with exacerbated proteinopathy due to impaired autophagic clearance.
Of most interest, the negative allosteric modulator of the RyR, dantrolene, restored lysosomal acidification and autophagic clearance of intracellular protein aggregates in animal models of AD, as well as in iPSC-derived neurons from AD patients.
Overall, both studies concur in suggesting an overactivation of RyR in AD and identifying a possible pharmacological means to circumscribe autophagic defects and associated protein accumulation. This agrees well with our previous work showing that, in murine models of AD, an endoplasmic reticulum calcium leak was observed that could be accounted for by Ryanodine receptor post-translational remodeling and that pharmacological stabilization of RyR or genetic rescue could reverse synaptic plasticity alterations and normalize AD-linked cognitive defects (Lacampagne et al., 2017).
The two recent papers describe very well-designed studies with complementary approaches raising similar conclusions. These represent a huge step toward a better understanding of the phenotypic alterations linking Ca2+ signaling perturbation and autophagic processes. Besides AD, this molecular cascade and its dysfunction could well account for process taking place more generally in other brain proteinopathies.
References:
Lacampagne A, Liu X, Reiken S, Bussiere R, Meli AC, Lauritzen I, Teich AF, Zalk R, Saint N, Arancio O, Bauer C, Duprat F, Briggs CA, Chakroborty S, Stutzmann GE, Shelanski ML, Checler F, Chami M, Marks AR. Post-translational remodeling of ryanodine receptor induces calcium leak leading to Alzheimer's disease-like pathologies and cognitive deficits. Acta Neuropathol. 2017 Nov;134(5):749-767. Epub 2017 Jun 19 PubMed.
University College Cork
It was very interesting to read these two papers which, for the first time, link increased RyR2-induced calcium release from the ER with endosomal-autophagic-lysosomal (EAL) defects in AD. Both the RyR2 and EAL systems become aberrant at very early stages of AD, preceding the development of Aβ and tau pathogenesis. These results present the exciting prospect that “normalizing” increased activation of RyR2 Ca2+ release in AD neurons could ameliorate defects in EAL systems.
It's been more than 20 years since we discovered defective and increased RyR2 activity in brains of people who had sporadic AD (Kelliher et al., 1999). Since then discoveries from these teams, and others, have highlighted hyperactivation of RyR2, and Ca2+ homeostasis defects, as early events that can be targeted to prevent AD pathogenesis and cognitive decline, and now we can add the EAL system to this list.
What do these studies mean for understanding and potentially targeting the RyR2/EAL system to ameliorate AD? Both papers focus on Familial AD (FAD) mouse models, providing novel data, but making the results difficult to interpret with respect to sporadic AD. Mustaly-Kalimi et al. employ human iPSC-derived neurons from AD patients with the A246E PS-1 FAD mutation, importantly revealing translation to human neurons. It would help substantially to determine whether key components of RyR-2-EAL dysfunction described in both papers—reduced vATPase subunits, lysosomal acidity, calcineurin-AMPK-ULK1 pathway and EAL defects—translate to iPSC-derived human neurons from sporadic AD patients or to human iPSC neurons expressing sporadic AD risk genes such as APOE-ε4. Furthermore, determining how these EAL systems link to aberrant RyR2 in the postmortem brains of individuals who had AD at different disease stages would be enlightening.
The protection against lysosomal defects shown by blocking increased RyR2 Ca2+ release in Mustaly-Kalimi et al. is interesting, as is the close coupling between increased RyR2-induced Ca2+ release and lysosomal alkalinization. This latter finding supports previous work showing PS-1 deletion/FAD causing PS-1 mutants deacidify lysosomes, and increase lysosomal Ca2+ release via TRPML1 endolysosomal Ca2+ channels (Lee et al., 1999).
In contrast, our recent work discovered that TRMPL1-induced Ca2+ release is down-regulated in iPSC-derived human cortical neurons expressing APOE-ε4 compared to other APOE isoforms, is accompanied by increased levels of lysosomal Ca2+, and that this occurs in the absence of lysosomal alkalinization (Somogyi et al., 2023). Together, these highlight differential pathological effects that may occur in the RyR2/EAL system in neurons in sporadic AD, without PS-1 alterations.
Interestingly, in cardiomyocytes, RyR2 on the sarcoplasmic reticulum (SR) forms stable complexes with TRPML1 in late endosomes and lysosomes (Thakore et al., 2020). This coupling enables spontaneous RyR2-induced “calcium sparks,” and related Ca2+-activation of K+ channels, which modulate vasoconstriction. Remarkably, in the absence of TRPML1 Ca2+ release, RyR2 can no longer induce Ca2+ sparks, leading to RyR2 hyper-activation, hyper-excitation, and cardiac arrythmias (Thakore et al., 2020), It would be exciting to determine if an analogous situation occurs in neurons in sporadic AD.
Links between heart and brain health allow the tentative hypothesis that defective RyR2/TRPML1 EAL systems in cardiomyocytes may also occur in dementia/AD. Importantly, we show EAL defects induced by reduced TRPML1 Ca2+ release in neurons can be remediated by small molecule synthetic TRPML1 agonists (Somogyi et al., 2023).
Both papers focus on Aβ and tau as targets of increased RyR2 activity-induced autophagic failure. However, moving beyond Aβ and tau as autophagic “cargo” should be illuminating. Interestingly, proteomic studies are beginning to identify mitochondria, synaptic, and ER proteins targeted by autophagy to regulate synaptic function and neuronal firing (Coughlan and Maday, 2023), which are likely to be important in AD pathogenesis via the RyR2-EAL system. In summary, these papers have discovered a novel pathway linked to AD pathogenesis and open the door to future discovery to better understand and target increased RyR-2-induced Ca2+ coupling to EAL pathogenesis at early disease stages of AD.
References:
Kelliher M, Fastbom J, Cowburn RF, Bonkale W, Ohm TG, Ravid R, Sorrentino V, O'Neill C. Alterations in the ryanodine receptor calcium release channel correlate with Alzheimer's disease neurofibrillary and beta-amyloid pathologies. Neuroscience. 1999;92(2):499-513. PubMed.
Somogyi A, Kirkham ED, Lloyd-Evans E, Winston J, Allen ND, Mackrill JJ, Anderson KE, Hawkins PT, Gardiner SE, Waller-Evans H, Sims R, Boland B, O'Neill C. The synthetic TRPML1 agonist ML-SA1 rescues Alzheimer-related alterations of the endosomal-autophagic-lysosomal system. J Cell Sci. 2023 Mar 15;136(6) Epub 2023 Mar 21 PubMed.
Thakore P, Pritchard HA, Griffin CS, Yamasaki E, Drumm BT, Lane C, Sanders KM, Feng Earley Y, Earley S. TRPML1 channels initiate Ca2+ sparks in vascular smooth muscle cells. Sci Signal. 2020 Jun 23;13(637) PubMed.
Coughlan ML, Maday S. Beyond housekeeping: autophagy regulates PKA signaling at synapses. Trends Neurosci. 2023 Mar;46(3):167-169. Epub 2023 Jan 28 PubMed.
Make a Comment
To make a comment you must login or register.