Calcium Disruptions Wreak Synaptic Havoc
Quick Links
In mouse models of Alzheimer's disease, before cognition fails, tau fibrillizes, or Aβ even begins to accumulate, calcium signaling goes awry in the brain as neurons release more of the cation from their intracellular stores than they should. At the Society for Neuroscience annual meeting, held November 15 to 19 in Washington, D.C., Grace (Beth) Stutzmann, Rosalind Franklin University, North Chicago, Illinois, revealed new data to explain how this calcium overload damages dendritic spines, diminishes synapses, and dampens their ability to release vesicles. Stutzman claimed that this imbalance impairs synaptic plasticity not only in single cells, but in the entire hippocampal network. She attributed the effects to excessive release of calcium through the ryandodine receptor (RyR) calcium channel. Stutzmann is developing drugs to restore normal ryanodine (RyR) receptor function, and introduced several compounds at the meeting.
The RyR receptor is a large, calcium-activated calcium channel that sits in the membrane of the endoplasmic reticulum (ER). People have three different isoforms. RyR1 predominates in skeletal muscle, while RyR2 and RyR3 are found in cardiac cells and neurons. Though inside cells, these receptors are sensitive to neurotransmission. During an action potential, calcium enters through ligand-gated channels on the plasma membrane and opens RyR receptors on the ER. The ER then releases some of its stored calcium through the RyR. Researchers, including Stutzmann, reported that RyR2 expression rises in AD mouse models and in people with the Alzheimer's (see Stutzmann et al., 2006; and Zhang et al., 2010). In 3xTg-AD mice, synaptic stimulation caused more calcium to flow through the RyR than in control mice (see image below) (see Aug 2009 news story on Chakroborty et al., 2009). In turn, the neurons were susceptible to synaptic depression (see Chakroborty et al., 2012). Stutzmann now finds that synapses begin to wither under this elevated calcium signaling, wearing down their ability to function.

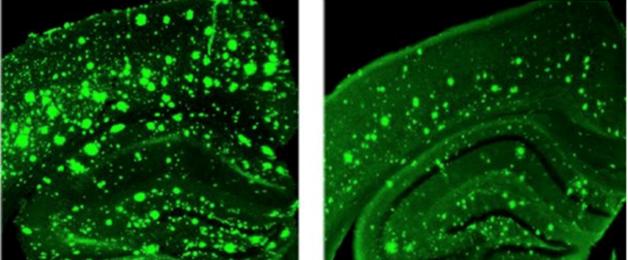
Overreaction:
In response to a synaptic stimulus, the dendrite of a 3xTg-AD mouse (right) releases three times as much calcium as a control dendrite. [Image courtesy of Grace (Beth) Stutzmann.]
Researchers in Stutzmann's lab compared 3- and 4-month-old wild-type to age-matched 3xTg-AD mice, before they developed AD-related pathology or cognitive deficits. Calcium irregularities had already begun (see Stutzmann et al., 2006). Using electron and confocal microscopy to examine synapses between the CA3 and CA1 area of the hippocampus, the researchers found that CA1 dendrites sported fewer mushroom spines, a type important for encoding long-term memories. CA1 and CA3 neurons formed fewer intact synapses with each other. The protein-rich region called the postsynaptic density, which is responsible for interaction with the presynaptic cell, became smaller. Presynaptically, the pool of neurotransmitter vesicles poised for release was drained.
The group found that last result particularly interesting. Could the depleted vesicles explain the synaptic depression Stutzmann’s group had previously found in these neurons? In patch-clamp recordings, the group found that these CA3 neurons spontaneously released more neurotransmitter vesicles than wild-type CA3 neurons. Stutzmann believes this constant spitting out of vesicles depletes the store available for release when a stimulus arrives. Then, fewer vesicles are released, stimulating synaptic depression rather than potentiation.
Did all this happen because of the excess calcium from RyR2 receptors? To find out, the researchers treated 3xTg-AD mice with dantrolene, an allosteric modulator that dampens RyR activity. The compound normalized calcium signaling in 3xTg-AD and TASTPM mice (see Chakroborty et al., 2012). At SfN, Stutzmann showed that dantrolene returned to normal the number of spines, synapses, and spontaneous vesicle released.
Dantrolene reduced tau and Aβ pathology in the PS/APP transgenic mice (see image below). Reducing calcium in the cells may dial back the activity of tau kinases and BACE1, Stutzmann said. “The extent of the structural and functional changes due to calcium dysregulation was profound,” she told Alzforum. “I was surprised that these were all reversed by stabilizing calcium signaling.” While she has no behavioral data on these mice, other groups have reported that dantrolene prevents and reverses cognitive deficits (see Peng et al., 2012, and Oulès et al., 2012).
A Reduced Load: After four weeks of dantrolene treatment (right) in PS/APP mice, Aβ pathology falls by half in the hippocampus and overlying cortex compared to saline-treated controls (left). [Image courtesy of Grace (Beth) Stutzmann.]
Gregory Brewer, University of California Irvine, noted that synapse loss accounts for the lion’s share of cognitive defects in the disease. “Any handle we can get on the reasons for synapse loss gets us closer to the cause of the disease, and is an obvious link to memory impairment,” he said. Did the changes in spines, synapses, and vesicles in the 3xTg–AD mice correlate with Aβ and tau pathology? Stutzman plans to address this question in the future.
Network Trouble
To address whether the synaptic deficits affect transmission across neural networks, Stutzmann, with William Frost and Shreaya Chakroborty, incubated hippocampal slices with a lipid-soluble fluorescent dye that absorbs more light and then fluoresces more intensely when the voltage across the cell membrane becomes more positive. A high-speed camera taking more than 1,000 pictures per second visualized the voltage changes through the hippocampal network. In control slices, a first synapse-strengthening stimulus causes a subsequent pulse to propagate farther through the network and depolarize neurons for longer than the first; both the greater distance and time are evidence of potentiation. However, in slices from 3xTg-AD mice, the second pulse spread no farther and lasted no longer than the first. This suggests, for the first time, that with elevated calcium release, the propagation of signals through the hippocampus is blunted in space and time. “It means regions of the hippocampus are less effective in transmitting information and sustaining it as they should be,” Stutzmann said. Next, she will test whether treatment with a RyR2 modulator restores network transmission.
“This is a great new method to image hippocampal networks,” said Olivier Thibault, University of Kentucky, Lexington, noting that it visualizes depolarization across large areas of the brain.
Looking for drug candidates that enter the brain better and are more specific than dantrolene, Stutzmann worked with medicinal chemists at her university, who synthesized allosteric RyR2 modulators. Stutzmann introduced four of the compounds, CK013, CK017, RD14, and RD95. All four normalized calcium responses in hippocampal slices from 3xTg mice. They did not markedly change other calcium-dependent properties of neurons, such as maintenance of membrane potentials. Given to 3-month-old 3xTg mice over four weeks, CK013 and CK017 restored RyR-induced calcium release to control levels. CK013 reduced tau and Aβ pathology in the hippocampus by more than half when given to 6-month-old TgCRND8 mice. Ongoing studies are testing whether this treatment preserves synaptic function and structure, and cognition in mice, Stutzmann said.
“These new derivatives of dantrolene that are effective in the central nervous system have provided new and exciting possibilities of treating patients before symptoms are prominent,” wrote Gary Gibson, Weill Cornell Medical College, New York, to Alzforum. “The remaining challenge is to show this approach is effective in human tissues and at non-toxic dosages.” Dantrolene itself is used as a muscle relaxant and approved for the treatment of malignant hyperthermia, an inherited disease of rising body temperature and muscle spasms.—Gwyneth Dickey Zakaib
References
News Citations
Research Models Citations
Paper Citations
- Stutzmann GE, Smith I, Caccamo A, Oddo S, Laferla FM, Parker I. Enhanced ryanodine receptor recruitment contributes to Ca2+ disruptions in young, adult, and aged Alzheimer's disease mice. J Neurosci. 2006 May 10;26(19):5180-9. PubMed.
- Zhang H, Sun S, Herreman A, De Strooper B, Bezprozvanny I. Role of presenilins in neuronal calcium homeostasis. J Neurosci. 2010 Jun 23;30(25):8566-80. PubMed.
- Chakroborty S, Goussakov I, Miller MB, Stutzmann GE. Deviant ryanodine receptor-mediated calcium release resets synaptic homeostasis in presymptomatic 3xTg-AD mice. J Neurosci. 2009 Jul 29;29(30):9458-70. PubMed.
- Chakroborty S, Kim J, Schneider C, Jacobson C, Molgó J, Stutzmann GE. Early presynaptic and postsynaptic calcium signaling abnormalities mask underlying synaptic depression in presymptomatic Alzheimer's disease mice. J Neurosci. 2012 Jun 13;32(24):8341-53. PubMed.
- Chakroborty S, Briggs C, Miller MB, Goussakov I, Schneider C, Kim J, Wicks J, Richardson JC, Conklin V, Cameransi BG, Stutzmann GE. Stabilizing ER Ca2+ channel function as an early preventative strategy for Alzheimer's disease. PLoS One. 2012;7(12):e52056. PubMed.
- Peng J, Liang G, Inan S, Wu Z, Joseph DJ, Meng Q, Peng Y, Eckenhoff MF, Wei H. Dantrolene ameliorates cognitive decline and neuropathology in Alzheimer triple transgenic mice. Neurosci Lett. 2012 May 16;516(2):274-9. PubMed.
- Oulès B, Del Prete D, Greco B, Zhang X, Lauritzen I, Sevalle J, Moreno S, Paterlini-Bréchot P, Trebak M, Checler F, Benfenati F, Chami M. Ryanodine receptor blockade reduces amyloid-β load and memory impairments in Tg2576 mouse model of Alzheimer disease. J Neurosci. 2012 Aug 22;32(34):11820-34. PubMed.
Further Reading
Papers
- Chakroborty S, Briggs C, Miller MB, Goussakov I, Schneider C, Kim J, Wicks J, Richardson JC, Conklin V, Cameransi BG, Stutzmann GE. Stabilizing ER Ca2+ channel function as an early preventative strategy for Alzheimer's disease. PLoS One. 2012;7(12):e52056. PubMed.
- Bruno AM, Huang JY, Bennett DA, Marr RA, Hastings ML, Stutzmann GE. Altered ryanodine receptor expression in mild cognitive impairment and Alzheimer's disease. Neurobiol Aging. 2012 May;33(5):1001.e1-6. PubMed.
Annotate
To make an annotation you must Login or Register.
Comments
Weill Cornell Medicine/Burke Neurological Instiute
Changes in calcium dynamics in cells from patients who have or will get Alzheimer’s disease have been reported for about 30 years (i.e., it is a trait-dependent, not state-dependent, marker). Considerable data at many levels (cell signaling, electrophysiology, molecular biology) have left no doubt the changes occur, and have established plausible molecular mechanisms to explain the changes. There have been many national and international meetings on the subject. The lack of development of therapeutic approaches based on the calcium changes has dimmed enthusiasm for this approach. The two abstracts by Dr. Stutzmann outlining new derivatives of dantrolene that are effective in the central nervous system have provided new and exciting possibilities of treating patients before symptoms are prominent. Dantrolene has also been proposed to treat cell death in ischemia in culture. The field also knows that we have cured “Alzheimer disease” in mouse models many times. Thus, the remaining challenges are to show this approach is effective in human tissues at non-toxic dosages. Dantroline is used as a muscle relaxant and no mention of those responses are described. I look forward to the papers, and to efforts to test this strategy in patients.…More
Stutzmann nicely extended her studies supporting the calcium-overload hypothesis of AD by providing morphological evidence of spine and synapse loss between the CA3 and CA1 region of the hippocampus in a mouse model of AD. These losses were associated with hypersensitive calcium signaling mediated by the ryanodine receptor. The heightened calcium signaling was shown to cause the synapse loss by demonstrating its reversal after administration of a ryanodine receptor modulator, dantrolene. These findings support the concept of calcium dysregulation and synapse loss from overexpression of amyloid in the 3xTg-AD mouse model before detectable memory impairment.
Stutzmann and colleagues advance a novel therapeutic approach of tamping down the calcium signaling to reverse early synapse loss due to amyloid-induced calcium hyperexcitability. Mechanistically, this fits nicely with Gina Turrigiano's principle of synaptic scaling (see Turrigiano, 2008) and lends support to Michaela Gallagher's proposition that AD is caused by synapse loss due to hyperexcitability (see Bakker et al., 2013). However, Stutzmann's results do not rule out a related mechanism in which early elevated basal calcium levels in neurons create a ceiling effect for synaptic signaling that is upstream of the overactive ryanodine receptor (see Parihar and Brewer, 2010). A calcium ceiling would limit synapse plasticity and limit energy from mitochondrial dehydrogenase upregulation: The mitochondrial calcium levels may already be high enough to maximally stimulate the mitochondrial dehydrogenases, so that further metabolic demand will not result in the needed boost in NADH production to power the electron transport chain and produce more ATP. This possibly also contributes to synapse elimination.…More
References:
Turrigiano GG. The self-tuning neuron: synaptic scaling of excitatory synapses. Cell. 2008 Oct 31;135(3):422-35. PubMed.
Bakker A, Krauss GL, Albert MS, Speck CL, Jones LR, Stark CE, Yassa MA, Bassett SS, Shelton AL, Gallagher M. Reduction of hippocampal hyperactivity improves cognition in amnestic mild cognitive impairment. Neuron. 2012 May 10;74(3):467-74. PubMed.
Parihar MS, Brewer GJ. Amyloid-β as a modulator of synaptic plasticity. J Alzheimers Dis. 2010;22(3):741-63. PubMed.
Make a Comment
To make a comment you must login or register.