Compound Stimulates Lysosome, Clears Tau in Human Cells and Mice
Quick Links
In the latest attempt to rid neurons of toxic tau, researchers are trying to harness and improve cells’ own clearance mechanisms. Researchers led by Kenneth Kosik, University of California, Santa Barbara, used a small molecule called lonafarnib to stimulate lysosomes and clear phosphorylated tau from neurons derived from patients with frontotemporal dementia (FTD). Treating tau transgenic mice with this compound for 10 weeks reduced tau tangles in the brain and prevented behavioral problems. The results, published March 27 in Science Translational Medicine, suggest a new therapeutic approach for tauopathies, including frontotemporal dementia (FTD) and possibly Alzheimer’s disease.
- Neurons derived from FTD patients are low in the Rhes protein.
- Suppressing Rhes clears tau pathology in patient-derived neurons and mouse models.
- The drug reduces microgliosis and rescues behavioral deficits in mice.
“This is an elegant study that clearly demonstrates the potential role of cellular protein degradation mechanisms in regulating tau pathology,” wrote Adam Boxer, University of California, San Francisco, to Alzforum. “It would be of interest to consider lonafarnib, or another agent that works via a similar mechanism, for other tauopathies.” However, he pointed out that the translatability of findings in transgenic mouse and iPSC models to sporadic human tauopathies is uncertain.
First authors Israel Hernandez and Gabriel Luna started by asking which genes were differentially regulated in human tauopathies. Patients with a variety of FTD-causing mutations in the MAPT gene—such as P301L, G55R, and V337M—donated fibroblasts and the researchers measured gene expression by RNA-Seq, comparing expression to that in control cells. Three genes emerged as differentially regulated—NEK9, ZFP4, and RASD2. RASD2 in particular caught their eye. It codes for Rhes, a member of the Ras family of GTPases that transmit extracellular signals into cells. Rhes hangs out on the inner side of the plasma membrane of neurons. The protein was previously reported to activate autophagy (Mealer et al., 2014).
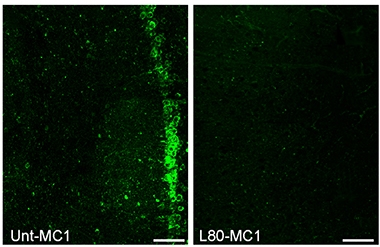
Keeping Tangles at Bay. In the hippocampi of 20-week-old rTg4510 mice, tau pathology (green), is drastically reduced by 10 weeks of lonafarnib treatment (right). [Courtesy of Science Translational Medicine/AAAS.]
In neurons differentiated from healthy iPSCs, Rhes levels slowly rose over time. In the patient-derived neurons, Rhes was downregulated compared with controls, and levels remained low. While not knowing why Rhes levels rose in healthy neurons, Kosik wondered if neurons from FTD patients downregulate Rhes as a protective response to mutant tau. Could decreasing it further help even more?
To explore that possibility, Kosik’s group turned to drugs initially developed for cancer called farnesyl transferase inhibitors. The Ras family all undergo a posttranslational modification called farnesylation, where a farnesyl transferase tags their C-terminal end with a branched 15-carbon group. The hydrophobic tail anchors the proteins to intracellular membranes. Without this anchor, Ras proteins get degraded, and this is the result of farnesyl transferase inhibition. One such inhibitor called lonafarnib seemed safe in Phase 1 cancer trials and it happens to cross the blood-brain barrier, meaning it might promote degradation of Rhes in the brain. Though it failed to slow tumor growth, it is being tested for progeria and hepatitis delta virus infection. Could lonafarnib protect cells from mutant tau?
To find out, the researchers treated neurons derived from patient fibroblasts with the compound. After nine days, Rhes levels fell considerably. So did levels of phosphorylated tau (p-tau) and sarkosyl insoluble tau, which goes on to become tangles. This prompted Hernandez and colleagues to try the drug in the rTg4510 transgenic mice, which express human tau carrying a P301L mutation. These mice develop tangles in the hippocampus, amygdala, entorhinal cortex, and cerebral cortex by 16 weeks, and about 60 percent of hippocampal neurons in CA1 die at about 22 weeks.
The researchers gave 10-week-old rTg4510 mice the drug by oral gavage, five days on and five days off, until they were 20 weeks old. Untreated animals preferred to sit apathetically in their cages, without, for example, building nests. When they did move, they often ran in circles. Treated mice, on the other hand, displayed few of these abnormal behaviors, nesting and moving around more like non-transgenic littermates. On histopathologic examination, the treated animals had a third to a half of the tangles seen in the hippocampi and cortices of untreated mice (see image above). Treatment also prevented brain atrophy typical for these transgenics, while reducing microgliosis in the hippocampus and tempering astrogliosis in the cortex. The authors noted that the drug did not work in 20-week-old mice, suggesting that once pathology had taken hold, lonafarnib was ineffective.
Was lonafarnib affecting tau clearance by suppressing Rhes? Modifying Rhes directly in 10-week-old transgenic mice, by silencing it with a small interfering RNA, similarly reduced tau pathology at 20 weeks, suggesting Rhes at least partly mediated the effects of the lonafarnib treatment.
To delve deeper, Kosik collaborated with Ana Maria Cuervo, Albert Einstein College of Medicine, New York. They found a dose-dependent increase in autolysosomes and in the clearance of tau from wild-type mouse fibroblasts and neuroblastoma N2A cells treated with lonafarnib. Substrates were more efficiently delivered to lysosomes, their degradation products disappeared faster, and the organelles were more readily degraded. The results suggest the drug was revving up the autophagy system, specifically improving lysosome efficiency.
Kosik’s lab is now focused on mapping the pathway that connects Rhes with the lysosome. He suggested that preventing its farnesylation somehow alters the signaling that regulates the lysosomal system. Learning more about this pathway could reveal other possible intervention points, he said.
Boxer said it was problematic that the drug only worked before tangles had developed. “While theoretically it might be possible to adopt such a prevention strategy in human MAPT mutation carriers, strictly speaking we don’t know when tau pathology begins in these individuals,” he wrote. “That makes it very challenging to know when to intervene.”
Going forward, Kosik would like to measure lonafarnib’s target engagement in the human brain. However, he said the drug’s sponsor, Eiger Pharmaceuticals, is reluctant to work with him. The company did not respond to a request from Alzforum for comment. Kosik is also testing similar farnesyltransferase inhibitors.
The strategy of lysosomal stimulation might offer an advantage over tau immunotherapy, which depends heavily on finding the right protein conformation to target, Kosik said. Since it is unclear which forms and phases of tau are toxic, researchers may choose the wrong target. “This [lonafarnib] strategy takes advantage of the cell’s own mechanism for clearing pathological tau and will help us get one more approach on the table,” Kosik told Alzforum.—Gwyneth Dickey Zakaib
References
Research Models Citations
Mutations Citations
Paper Citations
- Mealer RG, Murray AJ, Shahani N, Subramaniam S, Snyder SH. Rhes, a striatal-selective protein implicated in Huntington disease, binds beclin-1 and activates autophagy. J Biol Chem. 2014 Feb 7;289(6):3547-54. Epub 2013 Dec 9 PubMed.
Further Reading
Papers
- Mealer RG, Subramaniam S, Snyder SH. Rhes deletion is neuroprotective in the 3-nitropropionic acid model of Huntington's disease. J Neurosci. 2013 Feb 27;33(9):4206-10. PubMed.
- Scrivo A, Bourdenx M, Pampliega O, Cuervo AM. Selective autophagy as a potential therapeutic target for neurodegenerative disorders. Lancet Neurol. 2018 Sep;17(9):802-815. PubMed.
News
- Are Tauopathies Caused by Neuronal and Glial Senescence?
- Toxic Tau: Who Are You, and Where Are You From?
- A Proteomics Dive into Cause of Frontotemporal Dementia
- Viral Vectors Trigger Robust Tauopathy in Brain Slices
- Invasion of the Microtubules: Mutant Tau Deforms Neuronal Nuclei
- Another Reason to Catch Some Zzzs: Sleep Regulates Tau Release
Primary Papers
- Hernandez I, Luna G, Rauch JN, Reis SA, Giroux M, Karch CM, Boctor D, Sibih YE, Storm NJ, Diaz A, Kaushik S, Zekanowski C, Kang AA, Hinman CR, Cerovac V, Guzman E, Zhou H, Haggarty SJ, Goate AM, Fisher SK, Cuervo AM, Kosik KS. A farnesyltransferase inhibitor activates lysosomes and reduces tau pathology in mice with tauopathy. Sci Transl Med. 2019 Mar 27;11(485) PubMed.
Annotate
To make an annotation you must Login or Register.
Comments
UCSF
This is an elegant study that clearly demonstrates the potential role of cellular protein degradation mechanisms in regulating tau pathology. While it is encouraging that lonafarnib is already approved for use in humans for other indications and might therefore be repurposed for use in human tauopathy patients, as pointed out by the authors, the drug was only effective if started prior to the onset of tau pathology.
While theoretically it might be possibly to adopt such a prevention strategy in human MAPT mutation carriers, strictly speaking, we don’t know when tau pathology begins in these individuals, which makes it very challenging to know when to intervene. There are numerous other challenges to clinical trials in MAPT mutation carriers, including the rarity of these individuals and their clinical and neuropathological heterogeneity that is strongly related to which MAPT mutation is present.
It would be of interest to consider lonafarnib, or another agent that works via a similar mechanism, for other tauopathies such as AD, CBD, nfvPPA, PSP, or CTE, however, the translatability of findings in transgenic mouse and iPSC models to sporadic human tauopathies is uncertain.
A final challenge is the lack of target engagement and other pharmacodynamic biomarkers that can assess whether a drug such as lonafarnib is engaging its target in the human brain and exerting the same biological effects on protein degradation as seen in the preclinical models. I would advocate for increased focus on early identification of such target engagement biomarkers for new therapeutic entities targeting tau in preclinical studies such as this one, if there is a desire to rapidly translate the findings to the clinic.
Make a Comment
To make a comment you must login or register.