When Glial Clocks Fall Out of Sync, Inflammation Ensues
Quick Links
The circadian clock—the body’s molecular timekeeper—is best known for tuning our sleep, metabolism, digestion, and body temperature to the cycles of dark and light. Yet it controls other functions, as well. At the Society for Neuroscience meeting held November 3–7 in San Diego, researchers reported that activation of astrocytes and microglia in the brain is one of those. Disrupting the clock led to untimely inflammation that destroyed synapses. Researchers proposed that this phenomenon could exacerbate harm done by Alzheimer’s pathology, and that correcting circadian imbalances might protect neurons in the face of neurodegeneration.
- Mice missing the core clock protein, Bmal1, have runaway astrogliosis.
- Mice lacking the clock protein REV-ERBα have activated microglia and damaged synapses.
- A REV-ERBα agonist reduced neuroinflammation.
Previous studies led by Erik Musiek at Washington University in St. Louis have indicated that disruption of the circadian clock precedes cognitive symptoms in people with preclinical AD. In AD mice, knocking out the core clock protein, Bmal1, dampens the daily ebb and flow of Aβ in the hippocampus, hastening plaque formation (Feb 2018 news). Neuroinflammation is also known to wax and wane with the clock, as Musiek reported that knocking out Bmal1 throughout the mouse brain led to massive astrogliosis, neuroinflammation, and synaptic damage (Musiek et al., 2013). More recently, researchers got a similar, though less extreme, effect by deleting Bmal1 only in astrocytes. Not only did the cells become over-activated in the brain, they also no longer supported survival of stressed neurons in culture (Lananna et al., 2018).
At SfN, first author Brian Lananna presented unpublished findings extending this line of inquiry. He reported that in mice whose astrocytes express no Bmal1, astrocytes exhibited what has become known as an A1 astrocyte phenotype, revving up expression of a cocktail of pro-inflammatory genes. The researchers also noticed that expression of one gene, chitinase-3-like protein 1 (Chi3l1), was almost entirely snuffed out. This gene encodes YKL-40, a regulator of inflammation that is also an AD biomarker. YKL-40 levels rise in the plasma and CSF as disease progresses (Craig-Schapiro et al., 2010; Alzbiomarker YKL-40 (Plasma); Alzbiomarker YKL-40 (CSF)). In support of a link between Chi3l1 and the circadian clock, expression of the gene rose when the scientists knocked out two key regulators of Bmal1 expression, Per1 and Per2, in astrocytes. Chi3l1 expression rose during the day, while mice slept, and fell at night, while the nocturnal rodents scurried about their cages.
Lananna hypothesized that circadian control of Chi3l1 expression could explain why astrocytes become unhinged when their clock is disrupted. He generated Chi3l1 knockout mice to investigate. Those animals developed a similar astrogliosis phenotype as the Bmal1 knockouts. In response to the inflammatory instigator, lipopolysaccharide, astrocytes in Chi3l1 KO mice pumped out double the amount of pro-inflammatory cytokines, such as IL-6, as controls. Chi3l1 KO astrocytes also made poor neuronal nursemaids. When Lananna bathed cultured neurons in conditioned media from astrocytes, only media from normal astrocytes, not Chi3l1 KO astrocytes, bolstered neuron survival in the face of hydrogen peroxide stress. On the other hand, when the researchers grew neurons in media from astrocytes overexpressing Chi3l1, the cells gained extra protection.
Finally, Lananna asked whether Chi3l1 played a protective role in the brain of the APP/PS1 mouse model. When crossed to a Chi3l1 KO background, their hippocampi shrank more than did those of control APP/PS1 mice, despite similar Aβ burdens. Together, the findings suggested that astrocytic Chi3l1 dampens neuroinflammation and protects neurons from harmful stressors, including Aβ accumulation.
Following his talk, researchers asked Lananna more about Chi3l1 expression, and what explains increases in its protein product, YKL-40, with AD progression. He told the audience that according to the RNA-Seq analysis of the cerebral cortex carried out at the late Ben Barres’s lab at Stanford University, Chi3l1 was predominantly expressed in astrocytes, and not in other brain cells such as microglia or neurons. While little is known about its function in the brain, YKL-40 reportedly dampens inflammation in the lung by binding the IL-13 receptor (He et al., 2013). As to why it might increase as AD worsens, he suggested that, similar to anti-inflammatory cytokines like IL-10, YKL-40 might rise in step with the pro-inflammatory response to compensate for the damage the cytokines cause as disease progresses.
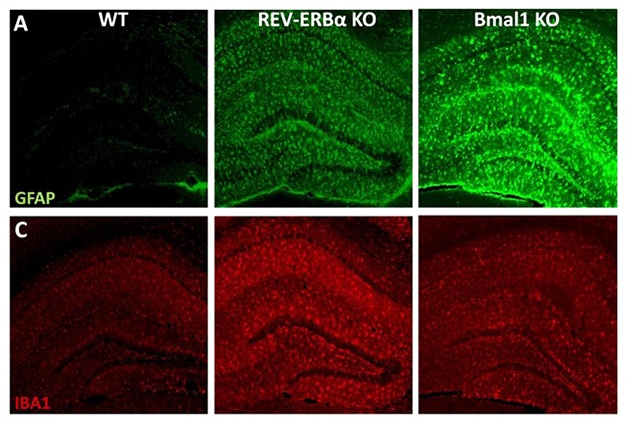
Glial Clocks Out of Sync. Knocking out REV-ERBα activates microglia (red, Iba1) and to a lesser extent, astrocytes (green, GFAP). Bmal1 KOs have more astrocytic than microglial activation. [Courtesy of Erik Musiek.]
While Bmal1 appears to promote Chi3l1 expression specifically in astrocytes, the master clock regulator also controls numerous other genes, many of which differ depending on cell type. To learn more about the pathways downstream of Bmal1, Musiek’s group also knocked out REV-ERBα. A nuclear orphan receptor, REV-ERBα is a key clock protein directly adjacent to Bmal1 in the circadian feedback loop. Bmal1 promotes expression of REV-ERBα, and in turn, REV-ERBα transcriptionally represses Bmal1. Knocking out Bmal1 reduces REV-ERBα expression by about 90 percent, while knocking down REV-ERBα elevates Bmal1 expression by only about 30 percent, Musiek told Alzforum.
At SfN, graduate student Percy Griffin reported that similar to Bmal1 knockout mice, the brains of mice lacking REV-ERBα were wracked with neuroinflammation and gliosis, and neurons in the hippocampus bore fewer synapses. However, the cause of the neuroinflammation appeared to arise from overactive microglia: expression of a host of microglial genes, including Iba1, TREM2, Ccl2, IL1b, and IL6, were elevated in the REV-ERBα knockouts. Complement proteins were also elevated.
Examining primary astrocytes and microglia in culture, Griffin found that knocking down REV-ERBα with siRNA did not alone spur astrocytes into activation. However, microglia did become activated by REV-ERBα knockdown, and when mixed with astrocytes, these microglia then instigated astrocytic activation as well. When the researchers collected media from these mixed glial cultures and added it to cultured neurons, they found that neurons grown in media from REV-ERBα knockdown glia were more sensitive to hydrogen peroxide stress than neurons grown in media from control glial cultures.
To home in on the specific role of REV-ERBα in microglia, Griffin is currently generating mice lacking REV-ERBα expression only in those cells. However, in the total REV-ERBα knockouts, Griffin already observed a strong disruption in circadian oscillations of microglial activation. In control mice, microglial expression of Iba1, a rough marker of activation, increases at night, while the animals are active. However, in the REV-ERBα knockouts, this rhythmicity is abolished, and microglia assume their nighttime levels of heightened activity 24-7. The researchers are currently looking at other activation markers and also assessing changes in microglial morphology throughout the day and night.
Also at SfN, researchers led by Rebecca Prosser at the University of Tennessee in Knoxville reported evidence that corroborated Griffin’s findings about the circadian oscillations of microglial activation, at least in the suprachiasmatic nucleus region of the brain, the home base of circadian rhythm research. There, similar to what Griffin saw in the hippocampus, microglia expressed more Iba1 at night. Examining microglial morphology, Prosser saw that, seemingly at odds with the finding that microglia expressed more Iba1 at night, they also stretched out their processes at that time, typically a sign of less activation. Prosser told Alzforum she was surprised by this, noting that interpreting morphological changes is not straightforward. Neurons in the suprachiasmatic nucleus are more active during the day—a rhythm that could influence microglia in this region.
Because microglia prune synapses—which can cause damage when in overdrive— Griffin and colleagues asked whether synapse numbers might also wax and wane in the mice, and whether that would change in REV-ERBα knockouts. Their preliminary findings, garnered from analyzing brain samples harvested at 10 a.m. (four hours after lights on) and 10 p.m. (four hours after lights out), indicate that synapse density is lowest when harvested at night. In REV-ERBα knockouts, this oscillation disappears, with synapses at a low level regardless of time of day, Griffin reported. The researchers have yet to connect this effect to oscillations in microglial activity or pruning. Still, they hypothesized that something about 24-7 microglial activation in REV-ERBα knockouts leads to synapse loss.
Could getting the clock back on track help stave off neuronal damage? The researchers proposed that as a nuclear receptor, REV-ERBα could make a therapeutic target. In fact, owing to its previously established roles in regulating metabolism in the liver, small-molecule agonists have already been developed. Griffin tested one of those, SR9009, in wild-type mice and in cultured cells (Solt et al., 2012). He reported that the drug dampened LPS-triggered neuroinflammation in the mouse hippocampus, and also promoted protection of stressed neurons in culture.
“The overall trend coming out of this research is that glial circadian clocks seem to regulate important processes in the brain, including neuroinflammation and synaptic pruning,” Musiek said, “If those go awry, that could potentially set the stage for cognitive impairment in the setting of disease pathology.”
While Musiek’s work suggests that circadian disruption could exacerbate damage caused by AD pathology, it is also possible that AD pathology messes with the clock. In support of this idea, researchers led by Inhee Mook-Jung at Seoul National University reported that in mice expressing human tau carrying the P301L mutation that causes frontotemporal dementia, tau pathology disrupted the oscillation of other key circadian functions, such as body temperature. The researchers tied this disruption to the degradation of Per2, a key clock protein that regulates Bmal1 expression.
Musiek told Alzforum that his lab is also investigating the relationship between tau pathology and circadian disruption. He added that just as the clock itself is controlled by tightly regulated molecular feedback loops, teasing out cause-and-effect relationships between disease pathology and circadian disruption will be a tall order.—Jessica Shugart
References
News Citations
Biomarker Meta Analysis Citations
Paper Citations
- Musiek ES, Lim MM, Yang G, Bauer AQ, Qi L, Lee Y, Roh JH, Ortiz-Gonzalez X, Dearborn JT, Culver JP, Herzog ED, Hogenesch JB, Wozniak DF, Dikranian K, Giasson BI, Weaver DR, Holtzman DM, Fitzgerald GA. Circadian clock proteins regulate neuronal redox homeostasis and neurodegeneration. J Clin Invest. 2013 Dec 2;123(12):5389-400. Epub 2013 Nov 25 PubMed.
- Lananna BV, Nadarajah CJ, Izumo M, Cedeño MR, Xiong DD, Dimitry J, Tso CF, McKee CA, Griffin P, Sheehan PW, Haspel JA, Barres BA, Liddelow SA, Takahashi JS, Karatsoreos IN, Musiek ES. Cell-Autonomous Regulation of Astrocyte Activation by the Circadian Clock Protein BMAL1. Cell Rep. 2018 Oct 2;25(1):1-9.e5. PubMed.
- Craig-Schapiro R, Perrin RJ, Roe CM, Xiong C, Carter D, Cairns NJ, Mintun MA, Peskind ER, Li G, Galasko DR, Clark CM, Quinn JF, D'Angelo G, Malone JP, Townsend RR, Morris JC, Fagan AM, Holtzman DM. YKL-40: a novel prognostic fluid biomarker for preclinical Alzheimer's disease. Biol Psychiatry. 2010 Nov 15;68(10):903-12. PubMed.
- He CH, Lee CG, Dela Cruz CS, Lee CM, Zhou Y, Ahangari F, Ma B, Herzog EL, Rosenberg SA, Li Y, Nour AM, Parikh CR, Schmidt I, Modis Y, Cantley L, Elias JA. Chitinase 3-like 1 regulates cellular and tissue responses via IL-13 receptor α2. Cell Rep. 2013 Aug 29;4(4):830-41. Epub 2013 Aug 22 PubMed.
- Solt LA, Wang Y, Banerjee S, Hughes T, Kojetin DJ, Lundasen T, Shin Y, Liu J, Cameron MD, Noel R, Yoo SH, Takahashi JS, Butler AA, Kamenecka TM, Burris TP. Regulation of circadian behaviour and metabolism by synthetic REV-ERB agonists. Nature. 2012 Mar 29;485(7396):62-8. PubMed.
External Citations
Further Reading
No Available Further Reading
Annotate
To make an annotation you must Login or Register.
Comments
No Available Comments
Make a Comment
To make a comment you must login or register.