DIAN and ADNI Data Say Familial and Sporadic AD Converge
Quick Links
Some ongoing prevention trials enroll people carrying familial Alzheimer’s disease mutations because they are destined to develop the disease on a predictable timescale. But will findings from these trials generalize to the vastly larger sporadic AD population? In other words, is dominantly inherited AD the same or different from sporadic AD? And what role does aging play? These questions spark debate whenever they come up, and researchers with the Dominantly Inherited Alzheimer Network set out to answer them once and for all. They are comparing the DIAN dataset to that from observational studies of late-onset disease. At the first Advances in Alzheimer’s and Parkinson’s Therapies Focus Meeting (AAT-AD/PD), held March 15–18 in Turin, Italy, John Morris and Randall Bateman at Washington University in St. Louis reported preliminary data from the study. The findings to date indicate that despite having different causes, familial and sporadic AD on the whole share the same pathophysiology and progression. The few distinctions between the diseases are mostly due to the older ages of people with sporadic AD, and the quixotic effects of particular AD mutations, the scientists noted.
- Researchers at DIAN and ADNI standardized their data sets for easy comparison.
- They found the two diseases shared almost identical pathology and progression.
- The findings suggest that treatments developed for familial AD would work for sporadic.
“We are cautiously optimistic that lessons learned from DIAN will translate to LOAD,” Morris said in Turin. This has big implications, because it suggests that a mechanism-based drug that proves its mettle in a DIAN study could be approved for sporadic disease.
One original aim of the DIAN project, which started enrolling in 2008, was to compare the features of symptomatic AD in dominantly inherited and the common late-onset sporadic populations, Morris noted (Nov 2008 news series). Both diseases are marked by altered Aβ42 homeostasis, but autosomal-dominant disease is caused by both an overproduction and impaired clearance of the peptide, while sporadic is believed to be due to impaired clearance (Jul 2010 conference news; Dec 2010 news; Jun 2013 news).

Merging Diseases.
Autosomal-dominant and sporadic AD have different triggers, but converge on the same pathophysiologic process. [Courtesy of Randall Bateman.]
An obvious comparison for the DIAN data set is ADNI, the longitudinal observational study of late-onset disease that started in 2004 (Mar 2005 conference news; Oct 2008 news series). DIAN’s initial protocols were based on ADNI’s to facilitate comparisons. However, over time, protocols in both studies diverged a bit. For one, ADNI replaced PiB amyloid imaging with florbetapir, while DIAN stuck with PiB. For another, the field has evolved, as in the move toward automated, more reliable cerebrospinal fluid assays (Aug 2015 conference news; Dec 2017 conference news). As a result, the data cannot be directly compared as originally gathered.
For this reason, researchers at DIAN and ADNI recently launched a joint effort to harmonize data from the two studies. They reprocessed all imaging scans using the centiloid scale, which renders a simple unit value for each scan and enables side-by-side comparisons between different tracers (Feb 2013 conference news; Nov 2014 news). They reanalyzed all CSF samples on the Roche Elecsys automated platform, and also compared Aβ isoforms by mass spectrometry, Morris said.
The researchers faced another challenge in comparing cohorts, namely the mean 30-year age difference between DIAN and ADNI participants. Because participants could not be matched by age, the researchers decided to match them instead by dementia severity. Progression to a CDR sum of boxes score of 1 indicated a person’s year of symptom onset, and served as the anchor point for the comparison. For some participants, these data were directly available, whereas for others who were already symptomatic when they enrolled, the researchers extrapolated back to the likely year of onset. For asymptomatic DIAN participants, researchers used the estimated year of onset based on their affected parent’s age at onset.

Faster Accumulation. People with a familial AD mutation (red lines) experience more aggressive amyloidosis than do people with sporadic AD (blue lines). [Courtesy of John Morris.]
The analysis comprised 559 ADNI participants and 302 mutation carriers in DIAN. Besides age, researchers noted a few demographic discrepancies between the cohorts. ADNI participants had more years of education than DIAN participants on average, and they were all from North America whereas DIAN is worldwide. In addition, ADNI, having been funded earlier, has more years of follow-up.
Despite these differences, most clinical and biomarker data were indistinguishable between the groups, Morris said. This is the main finding of the comparison study. Clinically, both diseases primarily involved defects in memory and executive function. Brain atrophy and hypometabolism occurred with similar patterns in each group. CSF biomarkers moved in the same ways, except the drop in CSF Aβ42 prior to symptom onset was steeper in DIAN than LOAD.
Ron Thomas of the University of California, San Diego, who heard the talks in Turin, agreed that the curves of biomarker change in autosomal dominant AD and LOAD resemble each other. “There is substantial similarity between the cascade of processes,” he wrote to Alzforum.
One difference cropped up in amyloid PET imaging, namely more deposition in the basal ganglia in people with AD mutations compared to sporadic AD. This has been reported previously, and occurs with specific presenilin 1 mutations (Jun 2007 news; Aug 2017 conference news). In addition, people with familial AD tended to accumulate a heavier amyloid burden than those with sporadic disease (see image above). Tau PET was only recently added to these studies and is still being analyzed, Morris noted.
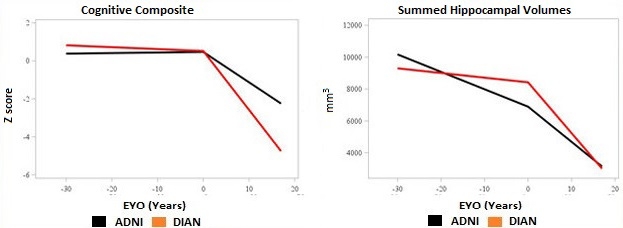
Earlier, Slower Decline. People with sporadic AD (blue lines) have some age-related decline before symptoms appear, but then progress more slowly than those with an AD mutation (red lines). [Courtesy of John Morris.]
The other discrepancies between the cohorts were attributable to age, Morris said. ADNI participants tended to have more brain shrinkage and cognitive decline prior to symptom onset than did DIAN participants, as might be expected in older adults. However, after symptom onset the pattern reversed, with DIAN participants experiencing steeper declines in brain volume and memory, Morris noted (see image above). “Symptomatic AD progresses more aggressively in dominantly inherited cases than in LOAD,” Morris said. This resembles the pattern seen in people with high cognitive reserve, who maintain cognitive function to a point of heavy amyloid load but then decline more quickly than people with low reserve.
Neuropathology data from participants who came to autopsy also exposed the aging component of LOAD. The younger DIAN participants developed pure AD pathology, whereas ADNI participants had comorbidities such as TDP-43 accumulation, argyrophilic grain disease, age-related tau astrogliopathy, hippocampal sclerosis, or vascular infarcts. Notably, in addition to amyloid plaques and neurofibrillary tangles, both DIAN and ADNI participants frequently developed α-synuclein deposits. Lewy bodies were present in 44 percent of ADNI cases and 54 percent of DIAN. This suggests that α-synuclein deposition is linked mechanistically to AD pathophysiology, Bateman noted. “There is important biology going on here that we do not understand,” he said in Turin.
Overall, dominantly inherited and sporadic AD are not identical, but they progress in the same way and have all the same core features, such that the diseases are difficult to distinguish clinically, Morris concluded. One indication of this is that a small fraction of people with late-onset AD turn out to have a familial mutation that went unrecognized. In fact, Bateman estimated that there might be 10 times as many mutation carriers with late-onset than early onset disease. In clinical practice, a 75-year-old person with symptomatic AD is much less likely to get tested for APP or presenilin mutations than a 50-year-old, but when scientists do look for mutations in aged cohorts, they tend to find some.
How will trials of LOAD deal with this? Samantha Budd Haeberlein of Biogen told Alzforum the company does not test for familial AD mutations when enrolling participants in aducanumab Phase 3 trials. Participants may be as young as 50, hence these trials may well include some carriers. When the trial data are analyzed, Biogen researchers will examine whether carriers responded to the treatment in the same way noncarriers did, Budd said.
DIAN researchers are comparing their dataset to other sporadic disease data, as well, including the National Alzheimer’s Coordinating Center (NACC) data set and the Adult Children Study at WashU, Morris noted. Participants in the latter are of a similar age to DIAN participants, removing one source of variation. Those data will be published soon. Virginia Buckles at WashU presented some preliminary findings at the 2016 Alzheimer’s Association International Conference in Toronto (Aug 2016 conference news).

Slow Clearance in ADAD/LOAD. Once amyloidosis sets in, mutation carriers (red line, left graph) clear Aβ42 as slowly as do people with sporadic late-onset AD (red line, right graph). [Courtesy of Randall Bateman.]
Also in Turin, Bateman presented data from stable isotope labeling kinetics (SILK) studies, which reveal the dynamics of Aβ production and clearance. SILK shows that Aβ isoform clearance slows by 2.5-fold from young adulthood to old age for everybody, regardless of mutation status, Bateman said. On the production side, mutation carriers make about 25 percent more Aβ42 than noncarriers throughout life. However, once amyloidosis starts, clearance slows identically in both carriers and noncarriers, Bateman stressed (see image above). Though the diseases start differently, their pathophysiology merges very early in the disease course, 15 or 20 years before symptoms emerge, he said. After symptoms appear, the disease lasts for about 10 years in either case.
The data suggest that treatments for dominantly inherited AD are likely to translate to LOAD. By way of an analogy, Bateman noted that statins work extremely well to prevent atherosclerosis and heart attacks in the rare cases of dominantly inherited familial hypercholesterolemia, and statins also work reasonably well in the general population in which this disease evolves more heterogeneously with an added aging component.
Thomas called the findings from these analyses important. “It would be a major step forward for AD research if autosomal-dominant AD can be considered as the same disease process, albeit accelerated, as LOAD,” he wrote.
Now if only the AD field had an effective drug. Several, such as Lilly’s solanezumab, Janssen’s BACE inhibitor, and Roche/Genentech’s crenezumab are currently being tested in both populations, allowing direct comparison of their effects.—Madolyn Bowman Rogers
References
News Citations
- DIAN: Registry for eFAD to Chart Alzheimer’s Preclinical Decade
- Honolulu: Wake-Up Call—Aβ Clearance, Not Production, Awry in AD
- Paper Alert: In Vivo Human Data Shows Reduced Aβ Clearance in AD
- In Familial AD, Aβ Production Up, Clearance Down
- Sorrento: ADNI Imagines the Future of AD Imaging
- As ADNI Turns Four, $64 Million Data Start Rolling In
- CSF Aβ Assays Remain Fickle: Robots to the Rescue?
- Automated CSF Tests: Check. Blood Tests: In the Works
- HAI—Standardizing Amyloid PET: The Centiloid Project
- Paper Alert: Centiloid Scale Aims to Unify Amyloid PET
- eFAD Research Surprise: In Mutation Carriers, Amyloid Starts in Striatum
- Data from DIAN Revise Familiar Biomarker Trajectories
- Monogenetic and Sporadic Alzheimer’s Disease: Same or Different?
Therapeutics Citations
Other Citations
Further Reading
News
- Amyloid Plaques’ Hold on Aβ42 Dampens Peptide’s Daily Rhythm
- In Familial Alzheimer’s, Tau Creeps into Cortex as Symptoms Show
- Data from DIAN Revise Familiar Biomarker Trajectories
- DIAN Longitudinal Data Say Cognition Goes Earlier Than Previously Thought
- Familial Alzheimer’s Gene Alters Children’s Brains
- API Biomarker Data Mirror DIAN’s, Support Progression Models
Annotate
To make an annotation you must Login or Register.
Comments
No Available Comments
Make a Comment
To make a comment you must login or register.