San Francisco: Tau—Time to Shine as Therapeutic Target?
Quick Links
At the “Tau and Tauopathies: Pathogenic Mechanisms” workshop held 28-30 March 2011 at the Gladstone Institute of Neurological Disease (GIND) in San Francisco (see Part 1), speakers voiced a sense of welcome relief, perhaps akin to foreigners enjoying the company of inhabitants of their native country in a faraway place. In this case, attendees did not all share a nation of origin, but, rather, bonded over a common research interest—the protein tau and its role in neurodegeneration. For years, the microtubule-associated protein has held the number two spot on the list of proteins implicated in Alzheimer’s disease, where amyloid-β has hogged the field’s focus. However, recent failures in AD clinical trials of amyloid-lowering approaches have left the field pondering. Researchers are asking whether they have gone after the wrong targets, relied too heavily on genetic models that may not capture sporadic disease, tested compounds in patients with confounding comorbidities, or whose disease is too advanced. Rising out of this contemplative frenzy is a growing sense that it’s high time to focus on tau—that this “runner-up” deserves more serious attention as a potential therapeutic target for AD and other tauopathies. This story will describe a slew of experimental treatment approaches, most at the cellular or rodent stage.
The theme of tau as an overdue target permeated a slide talk by Frank LaFerla of the University of California, Irvine. In a prior study, LaFerla’s group showed that setting off microglial inflammatory pathways worsened tau pathology in young triple-transgenic (3xTg-AD mice), and that blocking Cdk5 activation in the brain phagocytes could tone down these effects (ARF related news story on Kitazawa et al., 2005). The researchers found that tau pathology correlated with increased serum IL-1β in the transgenic mice, and wondered whether blocking signals through this inflammatory cytokine could possibly help. In experiments recently submitted for publication, the answer seems to be, yes. Unlike earlier approaches in the 3xTg-AD model that targeted tau but not did not improve Aβ pathology, treatment with anti-IL-1 receptor antibodies reduced both Aβ and tau arms of disease. It also lowered serum levels of key inflammatory cytokines and improved spatial memory in eight- to nine-month-old 3xTg-AD mice, LaFerla reported in San Francisco. To confirm and further explore the connection between tau and inflammation, the lab is crossing IL-1 knockout mice onto the triple-transgenic background.
LaFerla closed by recapping recent work his group did in collaboration with UC Irvine colleague Leslie Thompson, showing that four months of oral nicotinamide can boost cognition in 3xTg-AD mice (ARF related news story on Green et al., 2008). The treatment had no effect on amyloid-β levels or processing of its parent molecule, amyloid precursor protein (APP). As it turns out, the nicotinamide led to a dramatic reduction of Thr231-phospho-tau, as well as lower somatodendritic tau. The researchers have not yet looked for changes in axonal tau, LaFerla said.
Tau’s action in nerve projections is the focus of a different therapeutic approach under exploration by Kurt Brunden and coworkers at the University of Pennsylvania in Philadelphia. At the meeting, Brunden gave an update on his lab’s ongoing work with epothilone D—a brain-penetrant, microtubule-stabilizing compound that slowed axon degeneration and improved cognition when given to young PS19 tauopathy mice (Brunden et al., 2010; see also ARF conference story).
“It’s one thing to stop disease onset,” Brunden told the Gladstone audience. “But what about something more akin to treating patients who already have signs of disease?” Toward this end, Brunden reported preliminary data suggesting that epothilone had similar benefits for older PS19 mice in which tau pathology is well underway. Treated transgenics showed dose-dependent improvement in axonal dystrophy and microtubule density. They also had less hyperphosphorylated tau, as judged by AT8 immunohistochemistry using a system for Braak-like staging in mice (see Hurtado et al., 2010), and performed better in learning and memory tests. All told, the compound “given to PS19 mice either at or after onset of tau pathology improved a number of endpoints without safety or adverse events,” Brunden said. Bristol-Myers Squibb is planning a Phase 1 study of epothilone D, and would eventually like to test the compound in tauopathy patients, Brunden noted in an e-mail to ARF.
Grappling With the Basics
Other therapeutic approaches have taken shape as scientists ponder fundamental questions in the tau field. For instance, if tangles appear unnecessary for neuron death, and certain types of cellular dysfunction develop well before the tangles crop up (see Part 1), then what is it that makes tau neurotoxic? Some think post-translational modifications are key, while others place their bets on conformational changes. To Peter Davies of Albert Einstein College of Medicine, New York, an early leader in tau research, “both are probably true.” Davies presented recent data supporting each claim. In one study, he focused on c-Abl. The tyrosine kinase is fairly inactive in normal brain, but can phosphorylate tau, and associates with plaques and tangles in the brains of AD patients (Tremblay et al., 2010). In the March 2 Journal of Alzheimer’s Disease (Schlatterer et al., 2011), Davies and coworkers report on their new mouse with doxycycline-regulatable c-Abl activity in forebrain neurons. “If you express c-Abl in the forebrain, you get tau phosphorylation, cell death, and gliosis accompanying the cell loss,” Davies said. These data suggest that c-Abl may have a role in neurodegenerative disease, and could hold promise as a therapeutic target. Helping that case is the fact that compounds inhibiting this kinase (e.g., the cancer drug imatinib) are well tolerated in people.
Davies also reported preliminary results that speak to the second claim—that conformational changes are required for tau to turn toxic. Here, the researchers tested passive immunization for clearing tau pathology in JNPL3 transgenic mice expressing the P301L tau mutation that causes frontotemporal dementia. Starting at three months of age, the mice received four months of weekly intraperitoneal injections of a pan-tau monoclonal antibody, or a monoclonal (MC1) specific for a conformational tau epitope formed early in the aggregation process. In a pilot experiment, tau pathology dropped markedly in the MC1-treated group, as judged by total levels of insoluble tau in the brain, and pThr231 and pSer202 immunostaining in hippocampus.
“I’m stunned at how well this worked,” Davies said, though he noted that the mechanism remains unclear. Davies does not know what fraction of antibodies makes it into the brain, or whether an extracellular, prion-like form of tau may be involved. For now, the preliminary data suggest that the specificity of the antibody may be more important than affinity. At the 2010 Society for Neuroscience meeting in San Diego, California, Rakez Kayed of the University of Texas Medical Branch, Galveston, reported benefits from passive immunization of the same mouse (JNPL3) with a monoclonal antibody specific for tau oligomers. Kayed’s team gave eight-month-old transgenic mice a single hippocampal infusion of anti-tau oligomer antibodies, and found that the treatment cleared tau oligomers from CA1 and dentate gyrus neurons and improved the animals’ motor performance, compared to mock-injected controls (see ARF conference story).
Active and passive tau immunotherapy also helped tauopathy mice generated in the lab of Luc Buee at the University of Lille, France (Schindowski et al., 2006). Immunization with a phospho-tau peptide, or weekly injections of anti-phospho-tau antibodies, decreased tau pathology and prevented Y-maze memory deficits in this THY-Tau22 strain, which expresses mutant human tau (G272V and P301S) in the brain. It is notable in that these mice develop tau pathology as well as synaptic and cognitive impairments, but lack the motor symptoms that complicate behavioral testing of other tauopathy models. THY-Tau22 mice have clear learning deficits by nine to 10 months of age, some six months before neuronal loss is detected (Jeugd et al., 2011). Synaptic defects show up between nine and 12 months. The mice also have spontaneous seizures and gliosis that worsens with age, Buée reported. “In these mice, we can initiate long-term depression (LTD), like in normal mice, but cannot maintain it,” Buée said. LTD was restored in the THY-Tau22 mice by treating them with sodium selenate, a small compound that showed therapeutic potential in the pR5 and K3 tau mutant mouse models (Van Eersel et al., 2010).
Buée and colleagues also tested whether physical exercise could relieve tau pathology and cognitive woes in this tauopathy model. Starting at three months of age, the mice voluntarily ran about a kilometer each day for nine months. This regimen prevented behavioral defects in the Y-maze. “The running mice never develop this deficit,” Buée said. How was this possible? Buée and colleagues found no changes in transgene expression or in the gliosis that exists in this model in response to exercise; hence, disease persisted and exercise did not work through an anti-inflammatory pathway. Instead, the French scientists correlated the behavioral effect with decreased brain levels of phospho-tau at certain epitopes and with a preservation of cholinergic markers.
Interestingly, NPC1 and NPC2—cholesterol transport genes that are mutated in Niemann Pick’s disease—were upregulated after physical exercise, Buée said. Employing a viral gene therapy strategy that has reduced amyloid pathology in Alzheimer’s disease mouse models (Hudry et al., 2010), the Lille researchers injected into THY-Tau22 mice an adenovirus vector encoding neuronal cholesterol 24-hydroxylase. In preliminary studies, tau pathology was down and memory improved. Buée said he hopes to show more data in July at the International Conference on Alzheimer’s Disease in Paris, France.
Greg Cole of the University of California, Los Angeles, gave attendees yet another therapeutic strategy to chew on. This one involves feeding curcumin, a curry spice that Cole and colleagues previously showed can dissolve plaques and inhibit Aβ oligomerization in AD mice (ARF related news story on Yang et al., 2004). The researchers have now fed the spice to hTau mice for five months beginning at 14-15 months of age, when they already have tangle pathology, synaptic deficits, and cognitive impairment. Compared to untreated transgenic mice, hTau mice on the curcumin diet fared better in the Morris water maze, and expressed higher levels of excitatory synaptic markers NR2B and PSD-95, Cole reported. The treatment did not affect tangles, insoluble tau, or monomeric phospho-tau levels, but did suppress SDS-stable tau oligomers. The results are reminiscent of the cognitive recovery seen in tangle-bearing Tg4510 mice after tau transgene inhibition with doxycycline (ARF related news story on Santacruz et al., 2005), Cole wrote in an e-mail to ARF. The data “support tau oligomers as a target and curcumin as a pleiotropic drug capable of targeting pure tauopathy with late intervention,” he noted. Investigators at Jaslok Hospital and Research Centre in Mumbai, India, are recruiting AD patients for a Phase 2 trial of a highly absorbed, lipophilic curcumin formulation. Manufactured as a food supplement called LongVida by Verdure Sciences, Indianapolis, Indiana, this reportedly more bioavailable form of curcumin was originally developed by Cole and his wife, Sally Frautschy, also at UCLA.
Karen Duff of Columbia University, New York, spoke about a strategy that harnesses the macroautophagy system to tackle tauopathies. As shown on a poster by colleague Wai Haung Yu at the 2010 Society for Neuroscience meeting, eight-week treatment with the autophagy-promoting compound trehalose cleared tau aggregates and improved behavior in tauopathy mouse models with primarily motor (JNPL3 line) or cognitive (Tg4510 line) phenotypes (see ARF conference story). Preliminary functional magnetic resonance imaging (fMRI) done in collaboration with Scott Small, also at Columbia, suggests that trehalose can restore cerebral blood volume to normal in the Tg4510 mice, Duff said, noting that fMRI outcomes may be more applicable to human studies than rodent behavioral measures.
Another way of tapping intracellular degradation pathways to treat tauopathies came in a talk by Chad Dickey of the University of South Florida in Tampa. In addition to autophagy, cells use the ubiquitin-proteasome pathway to dispose of unwanted proteins, and heat shock proteins interact with molecules in both systems to help determine whether they will be dispatched in these ways. Dickey and others reported previously that inhibitors of the chaperone heat shock protein 90 (Hsp90) could promote degradation of mutated or hyperphosphorylated tau in mouse models of tauopathy (ARF related news story). In San Francisco, Dickey discussed his lab’s efforts manipulating another component of the chaperone system—Hsp70 proteins.
Collaborating with Jason Gestwicki at the University of Michigan, Ann Arbor, Dickey’s group screened for activators and inhibitors of Hsp70 ATPase activity and tested their hits in cell-based models and tau transgenic mice (Jinwal et al., 2009). Recently, the researchers have identified a new chemical scaffold, YM1, which is more potent and specific for the Hsp70 family than the earlier series of compounds. They have also found ways to tweak another variant, Hsp73 (aka Hsc70) to ready it for similar action. Normally, Hsp73 preserves tau (Jinwal et al., 2010). However, “we can make single mutations in this chaperone based on what we have learned using the Hsp70 ATPase inhibitors that force it to degrade tau instead,” Dickey explained in an e-mail to ARF. “With this information, we can home in on specific sites within Hsp73 that may be the most effective drug targets.” Targeting Hsp73 may be a good strategy because this variant is highly expressed in the brain and co-localizes with tau pathology, whereas other variants (e.g., Hsp72) do not.
Another potential therapeutic strategy centers around MSUT2, suggested Brian Kraemer of the University of Washington in Seattle. MSUT2 is the mammalian homolog of SUT2, a gene required for tau neurotoxicity in a transgenic worm model of tauopathy (ARF related news story on Kraemer et al., 2003). SUT2 overexpression caused the worms to rack up insoluble tau and lose neurons.
In a study published online in February, and appearing in print in this month’s issue of Human Molecular Genetics (Guthrie et al., 2011), the researchers found decreased levels of this protein in postmortem AD brain samples, but only in regions with tau pathology. Knockdown of MSUT2 in cultured tau-overexpressing human cells caused a dramatic drop in tau aggregation. All told, the data suggest that MSUT2 contributes to tau toxicity and aggregation, and that targeting this protein might be beneficial for tauopathies.
A big challenge for the field will be defining the tau forms that are most damaging, and isolating them from brains of patients. “We have to look in AD brain for these oligomeric species that a lot of people think are toxic,” Eva-Maria Mandelkow of the German Center for Neurodegenerative Diseases (DZNE), Bonn, Germany, said during the open discussion at the end of the meeting. Tau researchers may be taking their cues from the Aβ field, where the discussion changed once researchers started isolating oligomeric species from human AD brain after years of studying various synthetic or cell-based forms (see ARF related news story on Jin et al., 2011; ARF related news story on Shankar et al., 2008).
Offering a different way of thinking, Jürgen Götz, University of Sydney, Australia, questioned whether toxic tau species even exist in the human disease brain, after all. “Maybe the problem is tau elevation, whereby tau ends up in the wrong place and interacts with proteins it doesn’t normally associate with, preventing them from executing their physiological functions,” he said. Götz discusses evidence for this concept, dubbed the “tau axis hypothesis,” in a recent review with colleague Lars Ittner (Ittner and Götz, 2011).
Ultimately, once researchers finger the forms of tau posing the greatest threat in the human brain, it will be important to determine whether these toxic species can seed and propagate disease, or whether this requires different forms of aggregated tau. These ideas arise from work led by Michel Goedert at the MRC Laboratory of Molecular Biology in Cambridge, U.K., in collaboration with Florence Clavaguera and Markus Tolnay at the University of Basel, Switzerland. Their studies showed that misfolded tau can travel from cell to cell in a prion-like manner in the brains of tau transgenic mice (see ARF related news story on Clavaguera et al., 2009; see review by Goedert et al., 2010). Goedert also reviewed recent data he reported last fall at the 7th International Conference on Frontotemporal Dementias in Indianapolis, Indiana (see ARF conference story). In those experiments, mice that received brain extracts from people with tauopathies went on to develop pathological deposits that resembled the human disorder (see image below).
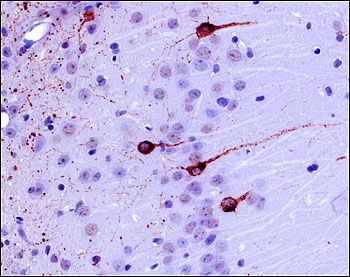
Filamentous tau deposits (labeled with AT100 antibody) in the CA1 region of the hippocampus in an ALZ17 tau transgenic mouse 12 months after injection with human AD extract. Image credit: Florence Clavaguera
If misfolded proteins propagate in this fashion in human disease, “it suggests that tau pathology may start in just one cell, maybe even following the misfolding of a single tau molecule,” Goedert said. “Over a period of time, it would then spread from this one cell to other cells. The unpredictable nature of these early events means that it will be difficult to predict with great precision who will develop a tauopathy.”
Though many questions remain unanswered, attendees left energized about the surge of recent tau developments highlighted at the meeting. Some also voiced caution about relying too heavily on mouse models, noting how transgenic mouse studies have at times infused AD researchers with misguided optimism about preclinical approaches. Nevertheless, “we were really able to drill down deep,” Mucke said at the meeting’s conclusion. The next GIND/DZNE meeting will take place in Bonn, probably in May 2012, where the drill will penetrate the topic of synaptic pathophysiology in neurodegenerative disease.—Esther Landhuis.
This concludes a four-part series. See also Part 1, Part 2, and Part 3. Read the PDF of the entire series.
References
News Citations
- San Francisco: Gladstone Institute Hosts Tau Powwow
- Inflammation Boosts Brain CDK5 Activity, Tau Phosphorylation
- Sirtuin Inhibitor Boosts Cognition, Reduces Phospho-tau
- Indianapolis: Clinical Trials a Ripple, Scientists Hope for a Wave
- San Diego: Tau Oligomer Antibodies Relieve Motor Deficits in Mice
- Curry Ingredient Spices Things Up by Blocking Aβ Aggregation
- No Toxicity in Tau’s Tangles?
- San Diego: Stimulating Autophagy Improves Symptoms in Mice
- Therapeutic Takedown: Hsp90 Inhibitors Tackle Tau
- Human Tau Is No Help to Worms
- Patient Aβ Dimers Sufficient for Tau, Neuritic Changes
- Paper Alert: Patient Aβ Dimers Impair Plasticity, Memory
- Traveling Tau—A New Paradigm for Tau- and Other Proteinopathies?
- Indianapolis: Dissecting the Pathways Behind Frontotemporal Dementia
- San Francisco: Making Tau Toxic—Post-translational Changes Galore
- San Francisco: Is Tau Reduction a Good Thing?
Paper Citations
- Kitazawa M, Oddo S, Yamasaki TR, Green KN, Laferla FM. Lipopolysaccharide-induced inflammation exacerbates tau pathology by a cyclin-dependent kinase 5-mediated pathway in a transgenic model of Alzheimer's disease. J Neurosci. 2005 Sep 28;25(39):8843-53. PubMed.
- Green KN, Steffan JS, Martinez-Coria H, Sun X, Schreiber SS, Thompson LM, LaFerla FM. Nicotinamide restores cognition in Alzheimer's disease transgenic mice via a mechanism involving sirtuin inhibition and selective reduction of Thr231-phosphotau. J Neurosci. 2008 Nov 5;28(45):11500-10. PubMed.
- Brunden KR, Zhang B, Carroll J, Yao Y, Potuzak JS, Hogan AM, Iba M, James MJ, Xie SX, Ballatore C, Smith AB, Lee VM, Trojanowski JQ. Epothilone D improves microtubule density, axonal integrity, and cognition in a transgenic mouse model of tauopathy. J Neurosci. 2010 Oct 13;30(41):13861-6. PubMed.
- Hurtado DE, Molina-Porcel L, Iba M, Aboagye AK, Paul SM, Trojanowski JQ, Lee VM. A{beta} accelerates the spatiotemporal progression of tau pathology and augments tau amyloidosis in an Alzheimer mouse model. Am J Pathol. 2010 Oct;177(4):1977-88. PubMed.
- Tremblay MA, Acker CM, Davies P. Tau phosphorylated at tyrosine 394 is found in Alzheimer's disease tangles and can be a product of the Abl-related kinase, Arg. J Alzheimers Dis. 2010;19(2):721-33. PubMed.
- Schlatterer SD, Tremblay MA, Acker CM, Davies P. Neuronal c-Abl overexpression leads to neuronal loss and neuroinflammation in the mouse forebrain. J Alzheimers Dis. 2011;25(1):119-33. PubMed.
- Schindowski K, Bretteville A, Leroy K, Bégard S, Brion JP, Hamdane M, Buée L. Alzheimer's disease-like tau neuropathology leads to memory deficits and loss of functional synapses in a novel mutated tau transgenic mouse without any motor deficits. Am J Pathol. 2006 Aug;169(2):599-616. PubMed.
- Van der Jeugd A, Ahmed T, Burnouf S, Belarbi K, Hamdame M, Grosjean ME, Humez S, Balschun D, Blum D, Buée L, D'Hooge R. Hippocampal tauopathy in tau transgenic mice coincides with impaired hippocampus-dependent learning and memory, and attenuated late-phase long-term depression of synaptic transmission. Neurobiol Learn Mem. 2011 Mar;95(3):296-304. PubMed.
- van Eersel J, Ke YD, Liu X, Delerue F, Kril JJ, Götz J, Ittner LM. Sodium selenate mitigates tau pathology, neurodegeneration, and functional deficits in Alzheimer's disease models. Proc Natl Acad Sci U S A. 2010 Aug 3;107(31):13888-93. PubMed.
- Hudry E, Van Dam D, Kulik W, De Deyn PP, Stet FS, Ahouansou O, Benraiss A, Delacourte A, Bougnères P, Aubourg P, Cartier N. Adeno-associated virus gene therapy with cholesterol 24-hydroxylase reduces the amyloid pathology before or after the onset of amyloid plaques in mouse models of Alzheimer's disease. Mol Ther. 2010 Jan;18(1):44-53. PubMed.
- Yang F, Lim GP, Begum AN, Ubeda OJ, Simmons MR, Ambegaokar SS, Chen PP, Kayed R, Glabe CG, Frautschy SA, Cole GM. Curcumin inhibits formation of amyloid beta oligomers and fibrils, binds plaques, and reduces amyloid in vivo. J Biol Chem. 2005 Feb 18;280(7):5892-901. PubMed.
- Santacruz K, Lewis J, Spires T, Paulson J, Kotilinek L, Ingelsson M, Guimaraes A, DeTure M, Ramsden M, McGowan E, Forster C, Yue M, Orne J, Janus C, Mariash A, Kuskowski M, Hyman B, Hutton M, Ashe KH. Tau suppression in a neurodegenerative mouse model improves memory function. Science. 2005 Jul 15;309(5733):476-81. PubMed.
- Jinwal UK, Miyata Y, Koren J, Jones JR, Trotter JH, Chang L, O'Leary J, Morgan D, Lee DC, Shults CL, Rousaki A, Weeber EJ, Zuiderweg ER, Gestwicki JE, Dickey CA. Chemical manipulation of hsp70 ATPase activity regulates tau stability. J Neurosci. 2009 Sep 30;29(39):12079-88. PubMed.
- Jinwal UK, O'Leary JC, Borysov SI, Jones JR, Li Q, Koren J, Abisambra JF, Vestal GD, Lawson LY, Johnson AG, Blair LJ, Jin Y, Miyata Y, Gestwicki JE, Dickey CA. Hsc70 rapidly engages tau after microtubule destabilization. J Biol Chem. 2010 May 28;285(22):16798-805. PubMed.
- Kraemer BC, Zhang B, Leverenz JB, Thomas JH, Trojanowski JQ, Schellenberg GD. Neurodegeneration and defective neurotransmission in a Caenorhabditis elegans model of tauopathy. Proc Natl Acad Sci U S A. 2003 Aug 19;100(17):9980-5. PubMed.
- Guthrie CR, Greenup L, Leverenz JB, Kraemer BC. MSUT2 is a determinant of susceptibility to tau neurotoxicity. Hum Mol Genet. 2011 May 15;20(10):1989-99. PubMed.
- Jin M, Shepardson N, Yang T, Chen G, Walsh D, Selkoe DJ. Soluble amyloid beta-protein dimers isolated from Alzheimer cortex directly induce Tau hyperphosphorylation and neuritic degeneration. Proc Natl Acad Sci U S A. 2011 Apr 5;108(14):5819-24. PubMed.
- Shankar GM, Li S, Mehta TH, Garcia-Munoz A, Shepardson NE, Smith I, Brett FM, Farrell MA, Rowan MJ, Lemere CA, Regan CM, Walsh DM, Sabatini BL, Selkoe DJ. Amyloid-beta protein dimers isolated directly from Alzheimer's brains impair synaptic plasticity and memory. Nat Med. 2008 Aug;14(8):837-42. PubMed.
- Ittner LM, Götz J. Amyloid-β and tau--a toxic pas de deux in Alzheimer's disease. Nat Rev Neurosci. 2011 Feb;12(2):65-72. PubMed.
- Clavaguera F, Bolmont T, Crowther RA, Abramowski D, Frank S, Probst A, Fraser G, Stalder AK, Beibel M, Staufenbiel M, Jucker M, Goedert M, Tolnay M. Transmission and spreading of tauopathy in transgenic mouse brain. Nat Cell Biol. 2009 Jul;11(7):909-13. PubMed.
- Goedert M, Clavaguera F, Tolnay M. The propagation of prion-like protein inclusions in neurodegenerative diseases. Trends Neurosci. 2010 Jul;33(7):317-25. PubMed.
Other Citations
External Citations
Further Reading
Papers
- Schlatterer SD, Tremblay MA, Acker CM, Davies P. Neuronal c-Abl overexpression leads to neuronal loss and neuroinflammation in the mouse forebrain. J Alzheimers Dis. 2011;25(1):119-33. PubMed.
- Jinwal UK, O'Leary JC, Borysov SI, Jones JR, Li Q, Koren J, Abisambra JF, Vestal GD, Lawson LY, Johnson AG, Blair LJ, Jin Y, Miyata Y, Gestwicki JE, Dickey CA. Hsc70 rapidly engages tau after microtubule destabilization. J Biol Chem. 2010 May 28;285(22):16798-805. PubMed.
- Guthrie CR, Greenup L, Leverenz JB, Kraemer BC. MSUT2 is a determinant of susceptibility to tau neurotoxicity. Hum Mol Genet. 2011 May 15;20(10):1989-99. PubMed.
- Leung KK, Barnes J, Modat M, Ridgway GR, Bartlett JW, Fox NC, Ourselin S, . Brain MAPS: an automated, accurate and robust brain extraction technique using a template library. Neuroimage. 2011 Apr 1;55(3):1091-108. PubMed.
- Brunden KR, Zhang B, Carroll J, Yao Y, Potuzak JS, Hogan AM, Iba M, James MJ, Xie SX, Ballatore C, Smith AB, Lee VM, Trojanowski JQ. Epothilone D improves microtubule density, axonal integrity, and cognition in a transgenic mouse model of tauopathy. J Neurosci. 2010 Oct 13;30(41):13861-6. PubMed.
- Belarbi K, Burnouf S, Fernandez-Gomez FJ, Desmercières J, Troquier L, Brouillette J, Tsambou L, Grosjean ME, Caillierez R, Demeyer D, Hamdane M, Schindowski K, Blum D, Buée L. Loss of Medial Septum Cholinergic Neurons in THY-Tau22 Mouse Model: What Links with tau Pathology?. Curr Alzheimer Res. 2011 Sep 1;8(6):633-8. PubMed.
News
- Indianapolis: Clinical Trials a Ripple, Scientists Hope for a Wave
- Therapeutic Takedown: Hsp90 Inhibitors Tackle Tau
- San Francisco: Gladstone Institute Hosts Tau Powwow
- San Francisco: Making Tau Toxic—Post-translational Changes Galore
- San Francisco: Is Tau Reduction a Good Thing?
- Inflammation Boosts Brain CDK5 Activity, Tau Phosphorylation
- San Diego: Stimulating Autophagy Improves Symptoms in Mice
- Sirtuin Inhibitor Boosts Cognition, Reduces Phospho-tau
- San Diego: Tau Oligomer Antibodies Relieve Motor Deficits in Mice
- Curry Ingredient Spices Things Up by Blocking Aβ Aggregation
- No Toxicity in Tau’s Tangles?
- Human Tau Is No Help to Worms
- Patient Aβ Dimers Sufficient for Tau, Neuritic Changes
- Paper Alert: Patient Aβ Dimers Impair Plasticity, Memory
- Traveling Tau—A New Paradigm for Tau- and Other Proteinopathies?
- Indianapolis: Dissecting the Pathways Behind Frontotemporal Dementia
Annotate
To make an annotation you must Login or Register.
Comments
No Available Comments
Make a Comment
To make a comment you must login or register.