New Ways to Target TREM2 Beg the Question: Up or Down?
Quick Links
Part 1 of 2
Microglia are the hot ticket in Alzheimer’s research these days, with a stream of studies showing that these cells can worsen or lessen pathology. In this balance, their receptor TREM2 acts as a pivot point, controlling a host of cell functions. Still, therapy-minded scientists remain unsure if it would be better to turn TREM2 up or down. So far, most research leans toward “up,” since pathogenic variants of TREM2 have reduced function. Now, work from Timothy Miller at Washington University in St. Louis casts a vote for “down.” In the July 6 Proceedings of the National Academy of Sciences, Miller and colleagues report that antisense oligonucleotides (ASOs) that squelch TREM2 expression halved plaque load in an amyloidosis mouse model. Surprisingly, belying other data, the drop in TREM2 seemed to activate microglial phagocytosis.
- Suppressing TREM2 with antisense oligonucleotides lowers plaque.
- Antibodies may block shedding of sTREM2 by nudging internalization.
- The right therapeutic strategy may depend on disease stage.
“There’s a brewing discussion about microglial states and whether they should be activated or inhibited, but the reality is that it is going to be disease-stage dependent and nuanced,” noted Kim Green at the University of California, Irvine. “We need better tools to address this, and these ASOs may help.”
It is also unclear what role the soluble sTREM2 fragment plays in microglial function, and what controls its shedding. In the June 25 Structure, researchers led by Alex Bullock at the University of Oxford, U.K., describe two antibody single-chain variable fragments (scFvs) that bind TREM2 and inhibit production of its soluble stub. The authors found that the antibodies dimerized before slowing TREM2 cleavage, and that they may act by stimulating cells to take up the receptor.
“The paper is a valuable contribution for the community, as it provides the first publication of the three-dimensional structure of TREM2-antibody complexes,” Kai Schlepckow at Ludwig-Maximilians University in Munich, Germany, wrote to Alzforum (full comment below). Like ASOs, scFvs may be valuable tools for TREM2 research, he added.
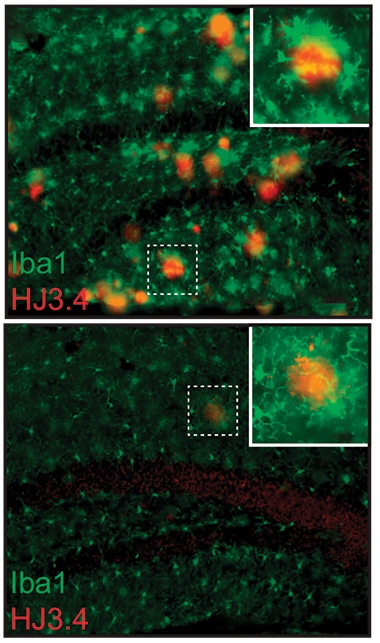
Less Active, Less Amyloid? Plaques (red) are numerous in the hippocampi of APP/PS1 mice (top). After TREM2 ASO treatment (bottom), plaques are sparse and microglia (green) more ramified, indicating a resting state. [Courtesy of Schoch et al., PNAS.]
TREM2 orchestrates microglial responses, boosting survival and phagocytosis to protect the damaged brain (Feb 2015 news; Jul 2016 news; Apr 2017 conference news). It also prods microglia to enter the disease-associated state seen around amyloid plaques in mice. When in this DAM state, the cells compact or perhaps even build plaques (May 2016 news; Sep 2017 news; Apr 2021 news). Several studies link TREM2 activation to fewer plaques but, confusingly, other studies report that TREM2 knockout lessens amyloidosis (Mar 2018 news; Dec 2014 conference news; Jay et al., 2017).
To disentangle this, first author Kathleen Schoch at WashU generated antisense oligonucleotides against mouse TREM2 and injected them into the ventricles of 10-month-old APP/PS1 mice. At this age, the mice have widespread plaques and highly activated microglia, as seen by Iba1 staining. A single jab with ASOs squelched TREM2 expression by 90 percent throughout the brain, and kept it low for longer than eight weeks.
One month after the injection, plaque load had dropped by half. Phosphorylated tau around plaques had fallen by a third. This p-tau may represent dystrophic neurites, the authors noted. Microglia around plaques looked less activated than in control mice, expressing less Iba1 and other pro-inflammatory markers, and having a more ramified shape (see image above).
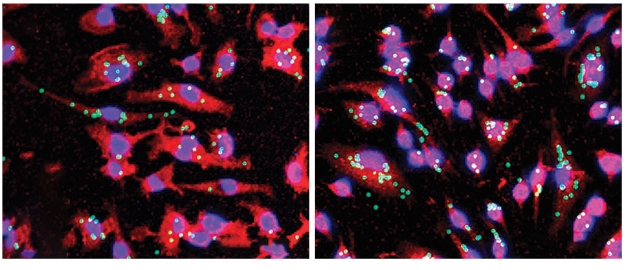
Better Eaters. Cultured microglia (red) expressing little TREM2 (right) consume more beads (green) than do controls (left). [Courtesy of Schoch et al., PNAS.]
How did ASOs produce this effect? At first, when the authors examined microglia one week after ASO injection, they saw a different picture. At this early point, microglia expressed more Iba1 and ApoE than in controls, suggesting the cells were activated. In keeping with this, isolated microglia from these mice gobbled up 50 percent more beads than those from controls, indicating enhanced phagocytic capability (see image above). The results suggest that knocking down TREM2 jolts microglia into a more phagocytic phenotype, spurring plaque cleanup. The subsequent drop in activation seen three weeks later may be a result of plaque clearance having quieted the microglia, Schoch wrote to Alzforum.
However, what the ASOs did depended on disease stage. ASOs injected at 4 months of age, before plaque deposition, or at 7 months, during plaque accumulation, had no effect on plaque pathology by 11 months. In other studies, too, manipulating TREM2 has different effects at distinct stages of disease (Jul 2018 conference news; Jan 2019 news; Apr 2020 conference news).
“Microglia are doing different things at different disease stages, and that needs to be considered when thinking about translation to humans, especially as pathology is occurring at different speeds and stages within the vastness of the human brain,” Green wrote to Alzforum (full comment below). Schoch agreed. “Our data would suggest that therapeutic targeting of TREM2 to alter microglial responses would be challenging,” she wrote.
The mice in this study had neither neurodegeneration nor cognitive decline at the age tested, so the study says nothing about whether inhibiting TREM2 to clear plaque would benefit these phenotypes. In addition, the mice do not accumulate tau, and some studies suggest TREM2 has opposite effects on amyloid and tau pathology (Oct 2017 news; Jul 2019 conference news; Jun 2020 news). Schoch plans to test TREM2 ASOs in mice with both pathologies.
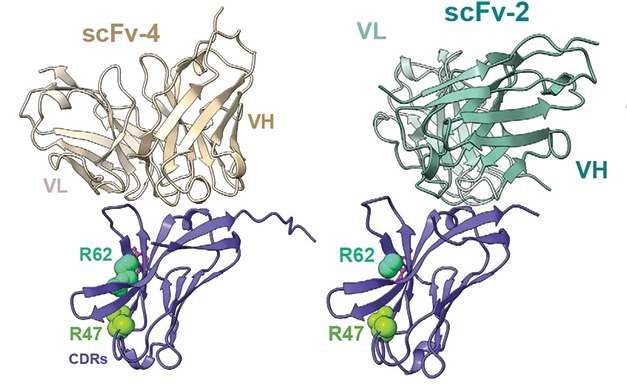
First Glance. Crystal structures of TREM2 (purple) bound to two single-chain variable fragments, scFv-2 (right) and scFv-4 (left). They bind TREM2’s immunoglobulin domain on the opposite side from the CDR regions that recognize ligands. [Courtesy of Szykowska et al., Structure.]
“While it remains unclear if targeting TREM2 is an appropriate therapeutic avenue, our study highlights the remarkable ability of microglia to transform the course of pathology in disease,” Schoch noted.
Most attempts to target TREM2 thus far have used antibodies, and two antibodies that activate the receptor are in clinical trials (May 2019 conference news; Mar 2020 news; Jun 2020 news).
Bullock and colleagues wanted to better characterize how TREM2 interacts with antibodies at a molecular level. To do this, first author Aleksandra Szykowska generated four scFvs against TREM2’s extracellular immunoglobulin-like domain, i.e., residues 19-131. Fragment 1 bound weakly and was set aside. Fragment 2 bound well enough to crystallize the complex and solve its structure (see image above). Alas, when added to cultured HEK293 cells expressing TREM2, scFv-2 did not affect sTREM2 shedding. Fragment 3 bound with higher affinity than scFv-2, and lowered sTREM2 by 22 percent. It dissociated quickly, however, so the crystal structure could not be solved.
Fragment 4 was the winner. It bound with high affinity and more specifically than did scFv-3, with less off-target binding. In culture, it lowered sTREM2 by 38 percent, meaning it was biologically active. The crystal structure of the complex showed that, like scFv-2, fragment 4 recognized an epitope on the other side of the immunoglobulin domain from the complementarity-determining regions (CDRs) that bind TREM2’s ligands. This implies that the antibody fragments would not interfere with ligand binding.
“Such binding sites would not be impacted by most AD risk variants, including R47H and R62H, suggesting a therapeutic potential of these scFvs to target TREM2 signaling,” noted Marco Colonna and Yun Chen at WashU.
Further probing revealed that scFv-3 and -4 affected TREM2 cleavage because they tended to form dimers. Purified monomeric preparations of fragments 3 and 4 had no effect, and fragment 2 did not dimerize, explaining why it was inert. “The need for scFv dimerization in order to inhibit TREM2 shedding is very much in line with what we have reported,” Schlepckow noted (Schlepckow et al, 2020). “It may be that both signaling and shedding of TREM2 involve receptor clustering,” Colonna and Chen agreed.
How do antibodies inhibit shedding? In cell culture, both scFv-3 and -4 caused cells to internalize TREM2, perhaps explaining how they prevent its getting clipped, the authors speculated. In contrast to these scFv fragments, the TREM2 antibodies in trials bind the stalk that connects the receptor’s immunoglobulin domain to its transmembrane portion. Thus, these antibodies may directly interfere with cleavage of sTREM2 (Aug 2017 news).
It is not completely clear if less TREM2 cleavage is good or bad in AD. Several studies have seen slower decline with high sTREM2, hinting cleavage is good (Jan 2016 news; Aug 2019 news). That said, is this because sTREM2 production reflects a high level of TREM2 signaling inside the cell, or because cleavage stops this signaling? If the latter, internalization of the receptor might accomplish the same thing. To boot, the soluble fragment on its own can also stimulate microglia, but again it is unclear if its effects are helpful or harmful (Feb 2017 news; Apr 2019 news).
Future studies may answer these questions. “We hope these additional tools will help elucidate the complex pathophysiology of TREM2 in AD,” Szykowska and colleagues wrote.
Lest a reader think microglia matter only in Alzheimer's, Part 2 of this series opens up a broader view on how these cells—through their ability to sculpt neuronal circuits—affect the brain in health and other diseases, particularly depression.—Madolyn Bowman Rogers
References
News Citations
- TREM2 Buoys Microglial Disaster Relief Efforts in AD and Stroke
- TREM2 Helps Phagocytes Gobble Up Aβ Coated in Antibodies
- New Evidence Confirms TREM2 Binds Aβ, Drives Protective Response
- Barrier Function: TREM2 Helps Microglia to Compact Amyloid Plaques
- ApoE and Trem2 Flip a Microglial Switch in Neurodegenerative Disease
- Microglia Build Plaques to Protect the Brain
- TREM2 Binds Aβ, Reprograms Microglia to Curb Plaques
- TREM2 Data Surprise at SfN Annual Meeting
- TREM2: Diehard Microglial Supporter, Consequences Be DAMed
- Without TREM2, Plaques Grow Fast in Mice, Have Less ApoE
- With TREM2, Timing Is Everything
- Changing With the Times: Disease Stage Alters TREM2 Effect on Tau
- TREM2, Microglia Dampen Dangerous Liaisons Between Aβ and Tau
- Boost or Block TREM2? Either Way, Therapy May Need Careful Timing
- Antibodies Against Microglial Receptors TREM2 and CD33 Head to Trials
- Paper Alert: Mouse TREM2 Antibody Boosts Microglial Plaque Clean-Up
- In Mice, Activating TREM2 Tempers Plaque Toxicity, not Load
- TREM2 Cleavage Site Pinpointed: A Gateway to New Therapies?
- TREM2 Goes Up in Spinal Fluid in Early Alzheimer’s
- In Alzheimer’s, More TREM2 Is Good for You
- Does Soluble TREM2 Rile Up Microglia?
- Cut Loose, Soluble TREM2 Beckons Microglia to Mop Up Plaques
- Not Just Alzheimer's: Microglia Sculpt the Brain in Health and Disease
Research Models Citations
Paper Citations
- Jay TR, Hirsch AM, Broihier ML, Miller CM, Neilson LE, Ransohoff RM, Lamb BT, Landreth GE. Disease Progression-Dependent Effects of TREM2 Deficiency in a Mouse Model of Alzheimer's Disease. J Neurosci. 2017 Jan 18;37(3):637-647. PubMed.
- Schlepckow K, Monroe KM, Kleinberger G, Cantuti-Castelvetri L, Parhizkar S, Xia D, Willem M, Werner G, Pettkus N, Brunner B, Sülzen A, Nuscher B, Hampel H, Xiang X, Feederle R, Tahirovic S, Park JI, Prorok R, Mahon C, Liang CC, Shi J, Kim DJ, Sabelström H, Huang F, Di Paolo G, Simons M, Lewcock JW, Haass C. Enhancing protective microglial activities with a dual function TREM2 antibody to the stalk region. EMBO Mol Med. 2020 Apr 7;12(4):e11227. Epub 2020 Mar 10 PubMed.
Further Reading
News
- Fall Flurry of Letters Kicks Up Dust Around TREM2
- TREM2 Mystery: Altered Microglia, No Effect on Plaques
- TREM2 Data Surprise at SfN Annual Meeting
- TREM2 Tidbits at AAIC: Genetics, Clinical Data
- United in Confusion: TREM2 Puzzles Researchers in Taos
- Unbiased Screen Fingers TREM2 Ligands That Promote Aβ Uptake
- Paper Alert: TREM2 Crucial for Microglial Activation
- Without TREM2, Microglia Run Out of Gas
- Model Morass? R47H Mutation Scuttles TREM2 Expression in Mice, Not People
- Down to Sex? Boy and Girl Microglia Respond Differently
- TREM2 Variants and CSF sTREM2 Levels Differ by Race
Primary Papers
- Schoch KM, Ezerskiy LA, Morhaus MM, Bannon RN, Sauerbeck AD, Shabsovich M, Jafar-Nejad P, Rigo F, Miller TM. Acute Trem2 reduction triggers increased microglial phagocytosis, slowing amyloid deposition in mice. Proc Natl Acad Sci U S A. 2021 Jul 6;118(27) PubMed.
- Szykowska A, Chen Y, Smith TB, Preger C, Yang J, Qian D, Mukhopadhyay SM, Wigren E, Neame SJ, Gräslund S, Persson H, Atkinson PJ, Di Daniel E, Mead E, Wang J, Davis JB, Burgess-Brown NA, Bullock AN. Selection and structural characterization of anti-TREM2 scFvs that reduce levels of shed ectodomain. Structure. 2021 Nov 4;29(11):1241-1252.e5. Epub 2021 Jul 6 PubMed.
Annotate
To make an annotation you must Login or Register.
Comments
University of California, Irvine
This is a compelling study demonstrating the power of ASOs. The authors show ASOs can potently target gene expression within microglia (especially for microglial-enriched genes), allowing for nuanced experiments that have been difficult to perform before.
Again, this study highlights that targeting microglia is sufficient to modulate many aspects of disease progression in mice, despite the pathological proteins being neuronally derived, hence it further validates microglia as a therapeutic target.
I also think it is important to highlight the timing effects of the treatment, which point to the fact that microglia are doing different things at different disease stages. That needs to be considered when thinking about translation to humans, especially as pathology is occurring at different speeds and stages within the vastness of the human brain.
It is not clear if it is the knockdown of Trem2 and the reversal of a DAM gene expression signature and association with plaques that may be beneficial, or if it is a transient early compensatory switch to a more plaque-degrading phenotype. This can be further explored and compared to the mechanisms of action of TREM2 activating antibodies, which also seem to be similarly beneficial.
Deutsches Zentrum für Neurodegenerative Erkrankungen (DZNE)
The paper by Szykowska et al. is a valuable contribution to the community, as it provides the first publication of the three-dimensional structure of TREM2 antibody complexes. Such studies are needed to identify and understand the molecular mechanisms of how therapeutic antibodies modulate TREM2 function.
The need for scFv dimerization in order to inhibit TREM2 shedding is very much in line with what we have reported in our 4D9 paper, where a Fab fragment of 4D9 failed to inhibit shedding in cell culture (Schlepckow et al., 2020).
Interestingly, the scFv antibody fragments reported in that paper do not bind to the stalk region as reported for previously published agonistic TREM2 Aβs (Schlepckow et al., 2020; Wang et al., 2020; Ibach et al., 2021), but rather to the Ig fold. It will be very important to investigate the effect of these scFvs on TREM2 downstream signaling, as previous studies have shown that agonistic TREM2 antibodies lead to increases in pSYK signaling, which again is dependent on the cross-linking activity of the antibody (Schlepckow et al., 2020; Ibach et al., 2021; Ellwanger et al., 2021).
A valuable feature of these antibody fragments in regard to future applications is the fact that their binding epitopes do not overlap with the putative ligand binding site in the Ig fold, or with the sites of the two most prominent LOAD-associated TREM2 variants at positions 47 and 62.
Future studies need to provide more insight into the mechanism of shedding inhibition, i.e., do the antibodies simply block access of ADAM proteases to the cleavage site or do they drive internalization leading to a reduction of cell surface TREM2 and thereby to a reduction of shed ectodomain, as discussed by the authors in their paper? Will that be dependent on where the antibody binds?
References:
Schlepckow K, Monroe KM, Kleinberger G, Cantuti-Castelvetri L, Parhizkar S, Xia D, Willem M, Werner G, Pettkus N, Brunner B, Sülzen A, Nuscher B, Hampel H, Xiang X, Feederle R, Tahirovic S, Park JI, Prorok R, Mahon C, Liang CC, Shi J, Kim DJ, Sabelström H, Huang F, Di Paolo G, Simons M, Lewcock JW, Haass C. Enhancing protective microglial activities with a dual function TREM2 antibody to the stalk region. EMBO Mol Med. 2020 Apr 7;12(4):e11227. Epub 2020 Mar 10 PubMed.
Ibach M, Mathews M, Linnartz-Gerlach B, Theil S, Kumar S, Feederle R, Brüstle O, Neumann H, Walter J. A reporter cell system for the triggering receptor expressed on myeloid cells 2 reveals differential effects of disease-associated variants on receptor signaling and activation by antibodies against the stalk region. Glia. 2021 May;69(5):1126-1139. Epub 2020 Dec 14 PubMed.
Wang S, Mustafa M, Yuede CM, Salazar SV, Kong P, Long H, Ward M, Siddiqui O, Paul R, Gilfillan S, Ibrahim A, Rhinn H, Tassi I, Rosenthal A, Schwabe T, Colonna M. Anti-human TREM2 induces microglia proliferation and reduces pathology in an Alzheimer's disease model. J Exp Med. 2020 Sep 7;217(9) PubMed.
Ellwanger DC, Wang S, Brioschi S, Shao Z, Green L, Case R, Yoo D, Weishuhn D, Rathanaswami P, Bradley J, Rao S, Cha D, Luan P, Sambashivan S, Gilfillan S, Hasson SA, Foltz IN, van Lookeren Campagne M, Colonna M. Prior activation state shapes the microglia response to antihuman TREM2 in a mouse model of Alzheimer's disease. Proc Natl Acad Sci U S A. 2021 Jan 19;118(3) PubMed.
Washington University School of Medicine
Washington University School of Medicine
Szykowska et al. have described four novel scFvs selected by phage display that bind to human TREM2, and the crystal structures of two with highest binding affinity/avidity. According to the reported structures of scFv-2- and scFv-4-TREM2 complexes, both scFv CDR-H3s bind to β strands A, F, and G of TREM2, where some hydrophobic residues may contribute to high binding affinity (e.g., Y104 and 105 on scFv-2 and Y102 and Y103 on scFv-4), although scFv-4 additionally binds to the C-C' loop on TREM2 (PDB: 6YYE, 6Y6C). This may explain the low affinity of scFv-1, since it lacks tyrosine at the N-terminal portion of CDR-H3.
Interestingly, the epitopes of both scFvs sit at the opposite site of the CDR loops of the Ig-like domain of TREM2. Importantly, such binding sites would not be impacted by most AD risk variants, including R47H and R62H, suggesting a therapeutic potential of these scFvs to target TREM2 signaling.
Previously, we demonstrated that an anti-mouse-TREM2 antibody (178) treatment has an anti-tumor effect similar to TREM2 deficiency (Molgora et al., 2020). This antibody may block TREM2 signaling after binding, in contrast to other anti-mouse/human-TREM2-activating antibodies tested in Alzheimer’s disease models including AL002a, AL002c, and 4D9 (Schlepckow et al., 2020; Wang et al., 2020; Ellwanger et al., 2021).
Although the details of molecular structures remain unclear, it is possible that epitope differences across those antibodies may contribute to opposite manipulations on TREM2 signaling. Therefore, it would be interesting to examine whether the similarity of the binding sites between scFv-2 and scFv-4 on the TREM2 Ig-like domain leads to similar functionality, and whether the extended interface between scFv-4 and C-C' loop of TREM2 completely alters the outcome.
Another interesting observation from this paper is that the protective activity against natural shedding of TREM2 tends to be dimer-specific. It is plausible that the reduction of TREM2 shedding requires scFv dimerization and is paralleled by complex internalization in their optimized experiments.
Two interesting questions await further demonstrations: (1) Does activation and shedding of TREM2 require oligomerization of the receptor? (2) Is the protection of shedding due to stabilization of TREM2 on the plasma membrane or promotion of antibody-TREM2 complex internalization? The second question is discussed by Szykowska et al., who suggest a role for internalization.
The first question is also worth discussing. According to the TREM2-PS structure (PDB: 6B8O, Sudom et al., 2018), the ligand binding requires another adjacent TREM2 molecule to form one complete binding pocket. TREM2 also forms a trimeric complex according to their crystal packing. In the high-resolution scFv-4/TREM2 complex structure (PDB: 6Y6C), it turns out that TREM2 molecules form dimeric complexes in this crystal packing. The putative ligand binding pockets (R47, R62) are toward the surface, and another risk variant, T96K, seems to be in the interface. Given that the oligomerization of scFvs is essential for protecting shedding, it may be true that both signaling and shedding of TREM2 involve receptor clustering and the interaction between scFvs and TREM2 are in multimeric states. The mutation on T96K may contribute to high-affinity self-dimerization that could explain the increase of signaling activation with this variant in our previous results (Song et al., 2017).
References:
Ellwanger DC, Wang S, Brioschi S, Shao Z, Green L, Case R, Yoo D, Weishuhn D, Rathanaswami P, Bradley J, Rao S, Cha D, Luan P, Sambashivan S, Gilfillan S, Hasson SA, Foltz IN, van Lookeren Campagne M, Colonna M. Prior activation state shapes the microglia response to antihuman TREM2 in a mouse model of Alzheimer's disease. Proc Natl Acad Sci U S A. 2021 Jan 19;118(3) PubMed.
Molgora M, Esaulova E, Vermi W, Hou J, Chen Y, Luo J, Brioschi S, Bugatti M, Omodei AS, Ricci B, Fronick C, Panda SK, Takeuchi Y, Gubin MM, Faccio R, Cella M, Gilfillan S, Unanue ER, Artyomov MN, Schreiber RD, Colonna M. TREM2 Modulation Remodels the Tumor Myeloid Landscape Enhancing Anti-PD-1 Immunotherapy. Cell. 2020 Aug 20;182(4):886-900.e17. Epub 2020 Aug 11 PubMed.
Schlepckow K, Monroe KM, Kleinberger G, Cantuti-Castelvetri L, Parhizkar S, Xia D, Willem M, Werner G, Pettkus N, Brunner B, Sülzen A, Nuscher B, Hampel H, Xiang X, Feederle R, Tahirovic S, Park JI, Prorok R, Mahon C, Liang CC, Shi J, Kim DJ, Sabelström H, Huang F, Di Paolo G, Simons M, Lewcock JW, Haass C. Enhancing protective microglial activities with a dual function TREM2 antibody to the stalk region. EMBO Mol Med. 2020 Apr 7;12(4):e11227. Epub 2020 Mar 10 PubMed.
Song W, Hooli B, Mullin K, Jin SC, Cella M, Ulland TK, Wang Y, Tanzi RE, Colonna M. Alzheimer's disease-associated TREM2 variants exhibit either decreased or increased ligand-dependent activation. Alzheimers Dement. 2017 Apr;13(4):381-387. Epub 2016 Aug 9 PubMed.
Sudom A, Talreja S, Danao J, Bragg E, Kegel R, Min X, Richardson J, Zhang Z, Sharkov N, Marcora E, Thibault S, Bradley J, Wood S, Lim AC, Chen H, Wang S, Foltz IN, Sambashivan S, Wang Z. Molecular basis for the loss-of-function effects of the Alzheimer's disease-associated R47H variant of the immune receptor TREM2. J Biol Chem. 2018 Aug 10;293(32):12634-12646. Epub 2018 May 24 PubMed.
Wang S, Mustafa M, Yuede CM, Salazar SV, Kong P, Long H, Ward M, Siddiqui O, Paul R, Gilfillan S, Ibrahim A, Rhinn H, Tassi I, Rosenthal A, Schwabe T, Colonna M. Anti-human TREM2 induces microglia proliferation and reduces pathology in an Alzheimer's disease model. J Exp Med. 2020 Sep 7;217(9) PubMed.
Make a Comment
To make a comment you must login or register.