Since the first Kloster Seeon meeting on β-secretases in 2013, a handful of BACE inhibitors have entered clinical trials and researchers have learned more about what these proteases do in development and adulthood. What’s the status of the field in the fall of 2016? Check out our coverage of the 2ndKloster Seeon meeting. Highlights include conditional BACE1 knockouts, updates on non-APP substrates cleaved by BACE, news about BACE’s role in dystrophic neurites, assays to measure off-target effects and—finally—the arrival of BACE1-selective inhibitors.
At 2nd Kloster Seeon Meeting, Renewed Optimism for Targeting BACE1
More than 100 BACE aficionados gathered September 25 near Munich for the 2nd Kloster Seeon Meeting on BACE Proteases in Health and Disease. Organized by Stefan Lichtenthaler, German Center for Neurodegenerative Diseases in Munich, and Robert Vassar from Northwestern University, Chicago, the meeting convened academic and industry leaders to take stock of the latest developments in BACE research and chart a path for further exploration. The meeting was a hit. It turns out trapping researchers in a converted monastery for two full days with access to only science, food, and a beer cellar leaves attendees begging for more. Highlights included reports of new conditional knockout mice that could lay bare the developmental and adult roles of BACE1, inhibitors selective for BACE1, updates on substrates both new and known, and some novel insights into the ramifications of BACE inhibition at the cellular level. Notably, there was more confidence than at the first Kloster Seeon meeting in 2013 that BACE inhibition just might end up working.
“This meeting is fantastic,” noted Pfizer’s Kelly Bales as she watched her fellow “monks for a day” struggle with one of the more vexing problems during after-dinner social time—how to knock down those bowling pins. “This is how all meetings should be organized,” was a refrain heard both above the din of the alley and in the meeting hall. Fun aside, a sense of camaraderie was palpable. “This is perhaps the only BACE meeting on the calendar where industry and academic researchers can share findings, discuss the challenges each group faces, and come to a mutual understanding,” noted Vassar. “We all have the same goal. We want to advance, and at end of the day, have [approved] drugs that treat or prevent AD in a safe and effective manner,” he told Alzforum.
Silence Is Golden?
A sense emerged from the meeting that the field was moving closer to that goal. A panel discussion between Ulf Neumann from Novartis, Basel, Switzerland, and Christian Haass, Ludwig Maximilians University, Munich, and moderated by Pierluigi Nicotera, who leads the German Center for Neurodegenerative Diseases, Bonn, was more tempered than the 2013 meeting, when academics voiced deep concern that pharma was heading down the same road with β-secretase inhibitors as they had with γ-secretase inhibitors. The latter not only hastened cognitive decline, but led to serious side effects, including skin cancer, that were attributed to interference with γ-secretase processing of Notch (see Nov 2012 news; Dec 2012 news).
Three years on, fears that neuregulin or some other β-secretase substrate might be the “Notch of the BACE programs” are fading. “There is now excitement in the BACE field that inhibitors may be safe enough for long-term treatment of AD,” said Lichtenthaler. This is partly because the retinal toxicity that plagued some early BACE inhibitors arose from off-target inhibition of cathepsin D, which newer BACE inhibitors avoid, and by the no-news-is-good-news silence from the ongoing clinical studies. AstraZeneca/Eli Lilly, Biogen/Eisai, Janssen/Shionogi, Merck, and Novartis have BACE inhibitors in Phase 2 or 3 trials, and so far there have been no reports of adverse events or intervention by the data safety review board to halt the trials, as there were with the γ-secretase inhibitors. Lynn Hyde from Merck, Kenilworth, New Jersey, said that as of June this year, 1,000 people had completed the 18-month Phase 3 EPOCH trial of their BACE inhibitor Verubecestat. Many at the meeting saw it as a hopeful sign that no serious safety issues have emerged from any of the ongoing trials.
Still, some concerns linger. Despite a long list of precautions and toxicity tests that Neumann listed as essential for any drug program, Haass wondered how γ-secretase inhibitors were ever allowed to move forward into the clinic. He worried that the same thing might happen again, to the detriment of the image and public support for Alzheimer’s research. Industry researchers agreed that with 20-20 hindsight, γ-secretase programs probably should not have moved forward. “Just as there are pressures in academia to publish, there can be pressure in industry to come up with something,” said Neumann, “and under that pressure the wrong decision can be made.” However, others noted that those decisions are not made in isolation—independent safety boards review data before and during trials. “We learned a lot from those trials,” said Samantha Budd from Biogen, Cambridge, Massachusetts. Others agreed, saying that the field is nowhere close to being in the same place with BACE inhibitors.
If academic scientists were concerned about industry’s rush to trial, then industry scientists were equally concerned about their academic colleagues’ rush to publish, sometimes with a penchant to exaggerate. Haass agreed. “Academics tend to overstate findings because they want to get papers in the best journals. This should stop immediately,” he said. Budd noted that if even one paper reports a potential side effect, then drug sponsors are obliged to test for it even if subsequent reports don’t bear out the original report. “This can increase the cost of trials, because unless you have proven [the side effect] is not in humans, you have to take that one publication into account. Toxicity tests do not stop with animal models, it continues all the way through the clinic,” she said.
That said, Lichtenthaler held that academic research not be dismissed too readily. Industry has criticized academia for publishing results that cannot be reproduced, and sometimes this is true, he acknowledged. “But the argument is also so fashionable these days, it is overdone.” While it is true that some phenotypes of BACE1 knockouts only emerge in certain animals, that does not mean we should just forget about them, said Lichtenthaler, who also holds a postion at the Technical University of Munich. “Background and strain difference come into play, and we don’t know which mouse best reflects the human population,” he said. Haass agreed that the current trend to simply view any preclinical study as being wrong is worrisome. “For example, Dale Schenk’s first AD vaccine study was completely right and highly reproducible,” he said.
Against this backdrop, how does the field today identify the side effects of BACE inhibition in mice that are likeliest to manifest in people? The researchers in Seeon appeared to rally around an initiative envisioned by Haass. “As we find new substrates, and new processing pathways for BACE, why don’t we set up a common platform to investigate them?” he asked, noting that any individual company would find it difficult to work on all of these areas by itself. “If we had a shared platform, we could finally come to concrete conclusions,” he said. Matthew Kennedy from Merck was all for this idea. He likened it to an initiative co-sponsored by the Michael J. Fox Foundation and industry to study inhibitors of the LRRK2 kinase. “Frankly, we’ve gained more from that in terms of data and knowledge than we have from the BACE field,” he said. Haass and Nicotera suggested that the EU would do well to fund this type of collaboration.—Tom Fagan
The 2nd Kloster Seeon Meeting on BACE Proteases in Health and Disease, held September 25-27 in Seeon near Munich, was propelled on an updraft of optimism that BACE inhibitors may work out to treat or prevent Alzheimer’s disease (see Part 1 of this series). And yet, even though no adverse events have stopped the ongoing Phase 2/3 trials, scientists are watching with bated breath because there is a long list of potential side effects that might still surface down the road. “My concern is that people may be on BACE inhibitors for decades,” said Robert Vassar, Northwestern University, Chicago. “We have to be aware of that and plan for long-term safety.” Stefan Lichtenthaler from the German Center for Neurodegenerative Diseases in Munich, who co-organized the meeting with Vassar, agreed. “We have not yet fully resolved whether BACE inhibition, per se, will lead to long-term toxicity,” he told Alzforum. The meeting tackled this issue head-on, such as with first reports on conditional knockouts of BACE1 in adult mice that enable a distinction between developmental versus adult roles of the protease. Other presentations explained how BACE processing of substrates beyond APP might affect synapses and plasticity.
Conditional KOs Debut
As industry leaders often point out, partially blocking BACE1 in adulthood and knocking it out in the embryo are wholly different things. While phenotypes have emerged in BACE1 embryonic knockouts that raise the specter of serious side effects of BACE inhibition (see table below), scientists have always wondered which of those may be rooted in developmental effects, and which truly arise in adult animals. To address this question, researchers in Vassar’s lab and in Riqiang Yan’s lab at the Lerner Research Institute, Cleveland, working independently, generated a total of four strains of conditional knockout models (cKOs). Both groups used the cre/lox system to pluck exon 2 out of the BACE gene, shutting down its expression.
Reported BACE1 Knockout Phenotypes. Scientists wonder which ones might be caused by chronic inhibition of BACE in older adults.
Vassar’s group engineered two different strains. In one they placed cre recombinase expression under control of the calmodulin kinase II promoter, which is active mainly in excitatory neurons in the forebrain. In the second, they ubiquitously expressed cre from the Rosa 26 promoter but made the knockout inducible with a tamoxifen-driven system. For his part, Yan’s group used the nestin promoter to drive cre expression in one strain and a tamoxifen-inducible cre driven by the ubiquitin promoter in a second. The nestin promoter turns on in neural precursors and neural stem cells, while the ubiquitin promoter is active in almost all cells. While these labs are still characterizing their mice, at Kloster Seeon they reported some notable phenotypes.
Vassar showed that unlike the embryonic BACE1 KOs, which struggle to wean and grow to be smaller than wild-type mice, the CamKII cKO mice weaned with ease and were close to normal size at three months old. They expressed no BACE1 in the cortex, a little in the hippocampus, and close to normal levels in the cerebellum. In keeping with this, the mice expressed higher levels of some BACE1 substrates in the cortex, including full-length APP, CHL1, and neuregulin 1. They had hardly any C99, the C-terminal part of APP left over after BACE cleavage.
Electrophysiologically, brain slices taken from year-old cKO mice showed normal long-term potentiation, a measure of synaptic plasticity. The mice navigated a Y maze normally, and they found the location of a hidden platform in the Morris water maze as well as control mice did in probe trials. Those are all good signs for BACE inhibitor programs, the researchers agreed. All the same, Vassar said these cKO’s may have a subtle problem with memory acquisition, as they took slightly longer to reach the platform in the first two days of learning trials.
More concerning perhaps is that, like the embryonic BACE1 knockouts, these CaMKII-driven cKOs suffered brain seizures. All six mice studied at one year of age had increased spike discharges detected by an EEG, and three of them had visible seizures as well. However, Vassar pointed out that CaMKII expression begins right after birth in these animals, making them a less faithful model of adult BACE inhibition than the inducible model.
In that vein, Vassar reported no seizure activity in the Rosa26 BACE1 knockouts when cre was induced at three, six, or nine months and the animals tested one to two weeks later. Again, histology showed that after tamoxifen, BACE1 was fully suppressed in the cortex but slightly less so in the hippocampus. These cKOs also had more APP, CHL1, and neuregulin in the brain. Myelination was normal, said Vassar. Hypomyelination is a phenotype reported for embryonic BACE knockouts. In Morris water maze probe trials, the Rosa26 BACE1 mice performed as well as controls did, but they, too, showed a slight delay in learning the position of the platform in the first day or two of training.
The males also may be weaker since they fell off a rotating rod more quickly than wild-type mice or their female siblings. Both male and female cKOs gained more weight than normal mice. This was totally unexpected since BACE1 embryonic knockouts are thinner. Vassar was perplexed by this and said it needs to be studied further. He was not sure that plumpness alone explains the males’ weaker grip on the rotarod. Hypomyelination or muscle spindle problems are also possible explanations for their apparent muscle weakness.
Yan reported that like the full embryonic BACE1 knockouts, his nestin-driven BACE1 knockouts were also smaller than normal mice. Because the nestin promoter turns on cre at around day 12 in the embryo, Yan concluded that early BACE1 expression in the neural lineage is necessary for normal growth. Yan found increased astrogenesis in the conditional knockouts as well, a phenotype that also happens in embryonic BACE1 knockouts.
In his inducible knockout, Yan and colleagues found reductions in BACE1 in the brain even before the animals were given tamoxifen, suggesting the leakage of the ubiquitin promoter that drives cre expression. This is a widely used promoter for cre-driven knockouts, but this leaky expression has never been reported before, said Yan. When they crossed these BACE1 cKO animal with 5xFAD mice, the offspring had dramatically fewer plaques at six months. This happened even without the addition of tamoxifen, in keeping with the leaky promoter hypothesis. Yan did not report exactly when this promoter begins to drive cre expression, but considered these observations highly important. “These data demonstrate that early inhibition of BACE1 will surely reduce amyloid deposition,” he told Alzforum. In addition, the BACE1 cKO had no sign of increased astrogenesis, which happens in the full embryonic knockouts. “This phenotype is more related to BACE1 function in early development rather than in the adult, indicating BACE1 inhibition will not induce this phenotype,” he concluded.
“While it’s early days in characterization, these conditional knockouts are important,” said Vassar. “Now we have tools to understand the physiological function of BACE in vivo in specific cell and tissue types, without the complication of developmental effects,” he said.
Lichtenthaler agreed. “If some phenotypes could be ruled as irrelevant for older people, we would not need to consider them potential side effects in clinical trials,” he said.—Tom Fagan
Dominguez D, Tournoy J, Hartmann D, Huth T, Cryns K, Deforce S, Serneels L, Camacho IE, Marjaux E, Craessaerts K, Roebroek AJ, Schwake M, D'Hooge R, Bach P, Kalinke U, Moechars D, Alzheimer C, Reiss K, Saftig P, De Strooper B.
Phenotypic and biochemical analyses of BACE1- and BACE2-deficient mice.
J Biol Chem. 2005 Sep 2;280(35):30797-806. Epub 2005 Jun 29
PubMed.
Further Reading
No Available Further Reading
BACE Inhibition and the Synapse—Insights from Seeon
Potential side effects remain a major concern for BACE inhibitor programs. Knocking out the gene for BACE1 in mice causes a variety of troubling phenotypes, not least being brain seizures (see Part 2 of this series). Among the more than 40 potential substrates for BACE1 in the proteome, several are likely relevant to seizure activity. At the 2nd Kloster Seeon meeting on BACE proteases, held September 25-27 near Munich, three stood out. They are seizure protein 6 (SEZ6), neuregulin 3, and MDGA1, a neuronal cell adhesion molecule linked to autism and schizophrenia.
Proteomic Analysis Lifts Lid on BACE Substrates.
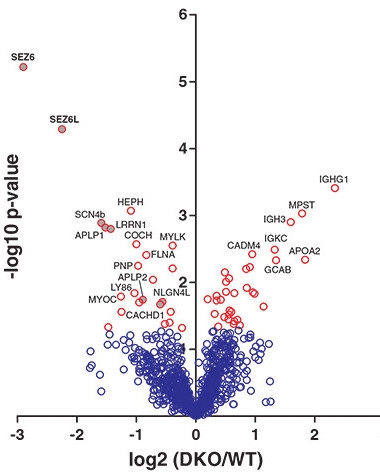
This “volcano” plot shows that levels of SEZ6 and SEZ6L ectodomains (x axis) are significantly (y axis) lower in CSF of BACE1/2 double knockout mice than in CSF from wild-type animals. [Courtesy of Pigoni et al., 2016.]
Researchers led by meeting co-organizer Stefan Lichtenthaler of the German Center for Neurodegenerative Diseases in Munich had previously identified the transmembrane protein SEZ6 as a potential BACE1 substrate (see Kuhn et al., 2012; Dec 2013 conference news). In Seeon, Lichtenthaler reported that shedding of SEZ6’s ectodomain plummeted eightfold both in BACE1 and in BACE1/2 knockout mice (see volcano plot at right) but not in BACE2 knockouts, fingering BACE1 as a major SEZ6 sheddase (Pigoni et al., 2016).
This held up in primates, as well. Analyzing rhesus monkey CSF samples after giving the animals a single dose of the BACE inhibitor MBI-4, the researchers found a reduction in SEZ6 ectodomain levels compared to samples taken after the same animal was given vehicle alone.
Could blocking the BACE1 cleavage of SEZ6 in adult animals be deleterious? To investigate, researchers in Jenny Gunnersen’s lab at the University of Melbourne, Australia, made SEZ6 conditional knockout mice. They expressed cre recombinase driven by a tamoxifen-inducible CamKII promoter in the animals, together with a floxed SEZ6 gene. In Seeon, Gunnersen showed that tamoxifen reduced SEZ6 expression in the CA1 region of the hippocampus and in the cortices of adult mice. This changed the animals’ behavior. They froze more easily in a contextual fear conditioning paradigm, suggesting they remembered better, even up to seven days after training.
Knocking out all three members of the SEZ6 family (i.e., SEZ6-like and SEZ6-like 2) seems to subtly affect memory as well (Miyazaki et al., 2006). In collaboration with Hiroshi Takeshima at Kyoto University, Japan, Gunnersen found that the triple KOs swam more slowly than controls, but learned to find the hidden platform in a water maze just as quickly. “This might be a sign that they initially learn slightly faster,” suggested Gunnersen. But in a reversal test, where the platform was moved to a new location, the mice took much longer to catch on. Gunnersen thinks cognition in these animals is rigid for some reason. This was also apparent in contextual fear conditioning. Here, too, the triple KOs seemed to learn better at first, freezing more readily when placed in a cage they associated with a foot shock, but while controls lost that memory over a few days, the triple KOs retained it.
It’s unclear what might underlie these effects. Gunnersen is looking for alterations in a number of brain regions, including the hippocampus and prefrontal cortex. As per electrophysiology, the paired-pulse ratio, which reflects synaptic vesicle release, was normal in the hippocampi of adult SEZ6 conditional knockouts, but excitatory currents were weaker, pointing to postsynaptic defects. In keeping with this, Gunnersen found that their synaptic spine heads were smaller. She said that soluble forms of SEZ6 may act like thrombospondin, which promotes synaptogenesis, and showed that secreted SEZ6 promotes excitatory synapse development similarly to thrombospondin in a model neuron culture system.
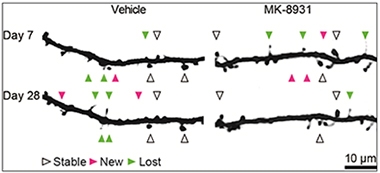
Spine Dynamics.
When given MK-8931 for three weeks, mice made fewer new dendritic spines and their spine density dropped. [Courtesy of Jochen Herms.]
Further evidence that healthy synapses might require SEZ6 came from Jochen Herms and colleagues at Ludwig Maximilians University, Munich. Previously, Herms had reported that BACE inhibitors impeded the formation of dendritic spines. Using two-photon microscopy to peer through a cranial window into the mouse brain, the researchers had recorded spine dynamics after administering high doses of either SCH1682496 or LY2811376, discontinued BACE inhibitors from Merck and Eli Lilly, respectively. In both cases the formation of new spines plummeted by half, and the overall numbers of spines fell 10 percent over a short period (see Nov 2014 news). In Seeon, Herms said that they have since seen the same effect with other BACE inhibitors, including MK-8931, which is being tested in Phase 2/3 clinical trials. After 21 daily doses of this drug, they observed a significant decrease in spine density due to impairment of new spine formation. Both spine formation and density bounced back once the drug was withdrawn. Interestingly, Herms does not see this effect in SEZ6 knockouts, suggesting this substrate may mediate the impact of BACE on spine turnover.
Herms also reported that knocking out SEZ6 caused postsynaptic deficits, as evidenced by reduced long-term potentiation. NB-360, another BACE inhibitor, had a similar effect, but not in SEZ6 knockouts, said Herms, again suggesting that BACE processing of SEZ6 plays a role in synaptic plasticity.
Interestingly, Lichtenthaler believes the BACE1 substrate MDGA1 may act at the postsynapse, as well. Researchers in his lab found more MDGA1 in whole brain extracts of BACE1 knockouts, suggesting BACE1 normally processes this membrane protein. In fact, Lichtenthaler reported that the BACE1 cleavage sites on MDGA1 and SEZ6 are very similar, and that neurons shed less soluble MDGA1 when BACE1 is knocked down or blocked pharmacologically.
What might be the consequences of blocking BACE1 cleavage of MDGA1? Others have shown that overexpressing MDGA1 prevents postsynaptic neuroligin-2 from interacting with presynaptic neurexin, thereby suppressing the formation of inhibitory synapses (see Pettem et al., 2013; Lee et al., 2013). If blocking BACE1 caused MDGA1 to accumulate, the effect might be the same. In keeping with this, Lichtenthaler’s group found that the BACE inhibitor C3 reduced the amount of the vesicular GABA transporter, VGAT, in mouse brain. GABA is the major inhibitory neurotransmitter in the central nervous system, and VGAT localizes to GABAergic synapses.
Evidence that BACE plays a role at the other side of the synaptic divide—the presynapse—came from Carmen Birchmeier, at the Max-Delbrück Center for Molecular Medicine, Berlin. Thomas Muller in her lab found that BACE1 not only cleaves neuregulin 3 (Nrg3), as had been reported by others (see Hu et al., 2008), but also stabilizes it on the neuron surface. This came as a surprise to Birchmeier and to others at the meeting, since many scientists have come to think of BACE cleavage as synonymous with ectodomain shedding. In fact, Birchmeier had previously found that BACE1 processed an isoform of neuregulin1 (Nrg1) in this manner and that this shedding was necessary for the proper formation of muscle spindles (Cheret et al., 2013). She reported that the protease, together with other sheddases, releases the Nrg1 extracellular domain, which then goes on to bind cell surface receptors (see July 2013 news). In Seeon, Birchmeier reported that BACE1 seems to cleave Nrg3 only at one site, which means the extracellular domain remains attached to the cell.
Birchmeier showed that Nrg3 occurs at synapses that express ErbB4. Specifically, she said Nrg3 in excitatory presynapses binds ErbB4 in postsynaptic membranes on inhibitory neurons. In fact, while Nrg3 knockout mice seem normal at first blush, they are a little hyperactive, but less impulsive, and mimic some symptoms of schizophrenia (see Loos et al., 2014; Hayes et al., 2016).
Could blocking or knocking out BACE1 have a similar effect to knocking out Nrg3? Cell culture experiments suggest yes. Birchmeier reported that BACE1 knockouts and cells expressing Nrg3 sans the extracellular domain had similar characteristics—namely, both had less ErbB4 in the postsynapse, and less synaptophysin in the presynapse. All told, Birchmeier thinks that by stabilizing Nrg3 at the cell surface, BACE1 helps strengthen excitatory synapses on inhibitory neurons.
Researchers were intrigued by these findings. On Lichtenthaler’s work, they noted how impressive the monkey CSF changes were after just one dose of BACE inhibitor. To the question of whether he would similarly analyze human CSF, Lichtenthaler said he would love to if he could get access to suitable samples. Others thought CSF “shedome” analysis might be valuable for companion diagnostics, a technique used to monitor drug efficacy in individual patients, asking if it could be used to test whether essential substrates of BACE1 were being spared by the drug at hand. Lichtenthaler considers it possible that cleavage of some substrates may be more strongly inhibited than others. “It might be interesting to see how far [CSF] Aβ falls while monitoring other substrates simultaneously. If they are not strongly reduced as well, we could then boost the dose of the inhibitor.”
Others were surprised that BACE seemed to have such a strong effect on the postsynapse. Robert Vassar, Northwestern University, Chicago, who co-organized the meeting with Lichtenthaler, said he had never even seen BACE there. Lichtenthaler agreed this was puzzling. “While many substrates are found on the postsynapse and BACE is seen on the presynapse I don’t think we fully appreciate where BACE1 cleavage occurs,” he said. “Not all BACE cleavage necessarily occurs in endosomes,” he added, suggesting the trans-Golgi might be a site for cleavage of postsynaptic membrane proteins. Vassar agreed, noting that BACE cleaves APP harboring the Swedish mutation on its way to the cell surface—it does not have to recycle through endosomes first.
Researchers further wondered if the soluble and membrane-bound forms of SEZ6 and MDGA1 have different functions and if the soluble forms can affect the presynapse. Gunnersen said the two forms of SEZ6 may have opposite functions, since overexpression of the membrane form inhibits dendritic branching, while the soluble form enhances it. She posited that the shed form interferes with protein-protein interactions of the membrane form and that this may help balance signaling events. She did not think that the soluble SEZ6 acts as a type of chemoattractant for the presynapse in development, though.
As for the presynapse, researchers wondered why Nrg3 levels fall if BACE does not cleave the protein. Birchmeier suggested that perhaps unprocessed Nrg3 never reaches the cell surface. “We are not sure yet,” she said, but noted that the loss of Nrg3 in the BACE1 knockouts is very obvious. Christian Haass, Ludwig-Maximilians University, Munich, wondered if Nrg3 and BACE1 translocated to the cell surface together as a dimer. Birchmeier thought that idea worth exploring.—Tom Fagan
Does BACE Drive Neurites into Dystrophy, Shorting Circuits?
Beta-secretase inhibitors are a leading therapeutic strategy being evaluated for Alzheimer’s disease, but as scientists discussed last month at the 2nd Kloster Seeon meeting on BACE Proteases in Health and Disease, there is still much to learn about the potential pitfalls of blocking these enzymes. Lo and behold, amid new data reinforcing worries of potential sides effects of BACE therapy (see Part 2 and Part 3 of this series), there were also some notable positive signs. Researchers reported how BACE inhibitors prevent dystrophic neurites from forming in animal models of AD, and might even help resolve dystrophies that are already there when treatment starts. Inhibitors also reined in neural hyperactivity in mice and restored long-range electrical synchronies that may be crucial for memory consolidation.
Dispatching Dystrophies
Researchers led by Jochen Herms at Ludwig Maximilians University, Munich, have used cranial windows in mice to monitor what inhibiting BACE does to neuritic dystrophy. The term denotes axons and dendrites swelling up near amyloid plaques. Packed as they are with BACE and APP, dystrophic neurites are potential sites for rampant Aβ production. To visualize the neurites, the researchers crossed APPPS1 transgenic mice with the VGlut1 venus mouse, which expresses a highly fluorescent form of green fluorescent protein in glutamatergic synapses.
Neuronal Dystrophy.
Glutamatergic presynaptic terminals and dystrophic axons (green) surround Aβ plaques (blue). [Courtesy of Jochen Herms.]
Herms showed exquisite images of dystrophic neurites that emerged beside plaques. These swellings grew over a period of weeks. Electron microscopy showed they were full of vesicles and proteins, including BACE and the lysosomal marker LAMP1. This could be a sign that late endosomes are getting stuck in the neurites and their proteins are not being properly degraded by the lysosomal system, said Herms. After about 12 weeks, the dystrophic neurites fizzled out. This most likely reflects outright destruction of the dystrophy, said Herms, but he also thinks that some neurons may be repairing dystrophies that lie furthest from plaques.
How would plaques and neurites respond to BACE inhibition? Herms treated the animals with NB-360, Novartis’ forerunner to CNP520, which is being tested in the Generation prevention trial of people who have two copies of the ApoE4 gene (see Aug 2016 conference news). In contrast to untreated mice, which accumulated new plaques and new dystrophies, the NB-360-treated animals made little of either. “The size of the dystrophic parenchyma stayed the same or grew only very slightly,” said Herms.
His talk spurred extensive discussion. Christian Haass, also of Ludwig Maximilians University, was intrigued that dystrophies only seemed to form after plaques had appeared, suggesting the plaques, and not a soluble form of Aβ, are the source of toxicity in this model. Herms agreed. “We have looked at synapses before plaques form and we do not see any pathology, so we have no evidence for soluble oligomeric toxic Aβ,” he said.
Robert Vassar, Northwestern University, Chicago, reminded the audience of David Holtzman’s two-photon work more than 10 years ago that demonstrated that anti-Aβ antibodies could shrink at least the smaller dystrophic neurites (see Jan 2005 news). “The difference here is that you are not removing plaques, so maybe the combination of an antibody and a BACE inhibitor would rescue the dystrophies,” he suggested. Herms said he thought BACE inhibitors by themselves might rescue some of the dystrophic neurites. “Those furthest from plaques may improve,” he said. “This may turn out to be a positive effect of BACE inhibition.” Vassar co-organized the Seeon meeting with Stefan Lichtenthaler from the German Center for Neurodegenerative Diseases in Munich.
Correcting Circuitry
Another upside of BACE inhibition might be that it could restore electrical connectivity in the brain, some scientists believe. Hyperactivity underlies silent seizures in some mouse models of AD, and possibly in people in the early stages of dementia as well (see Sep 2007 news; May 2012 news). Marc Aurel Busche from the Technical University of Munich previously reported that Aβ immunotherapy exacerbated hyperactivity in mice, possibly by unleashing soluble forms of Aβ from plaques (see Nov 2015 news). What would BACE therapy do?
Working in Arthur Konnerth’s lab, Busche has perfected live, two-photon calcium imaging to measure the activation state of neurons. He can simultaneously quantify the activity of individual cells and—thanks to a CCD camera that captures high-resolution images of whole regions of the brain at once—the activity of long-range neural circuits. Busche used these techniques to characterize the effect of NB-360 on APP23xPS45 transgenic mice (see Busche et al., 2008).
Tracing BACE. A green fluorescent protein tag allowed Tesco and colleagues to track BACE1 in axons. The somatodendritic protem MAP2B is labelled red. [Image courtesy of Selene Lomoio.]
Busche treated six- to seven-month-old mice for six weeks with inhibitor or placebo, then tested them in the Morris water maze. Then he imaged brain calcium by two-photon microscopy before sacrificing the animals to measure brain Aβ. The treated mice had fewer plaques and less soluble Aβ than controls, and navigated more quickly to the hidden platform in the water maze. They had almost no hyperactive neurons. Busche thinks NB-360 reduced hyperactivity by blocking Aβ production and allowing the mouse brain to fully expunge any Aβ oligomers that were loitering around the brain—Charlie Glabe’s OC antibody, which recognizes an epitope found in fibrillar oligomers but not soluble Aβ, failed to bind near plaques in the treated animals, but did in controls.
Finally, Busche reported that the BACE inhibitor cleared up disturbances in slow-wave oscillations. These patterns of long-range, synchronized firing spread over the entire brain when an animal is asleep or anesthetized. Scientists believe they help consolidate memories. In APP/PS mice, this synchrony breaks down as oscillations between the frontal and occipital cortices become uncoupled (see Busche et al., 2015). Camera-based calcium imaging of whole brain hemispheres revealed that NB-360 restored this coupling and that this restoration correlated with improved learning and memory.
Overall, Busche said he thinks that Aβ leads to hyperactivity, which breaks long-range circuits, which in turn leads to memory impairment. He suggested using functional MRI or EEG recordings to identify people with long-range circuity problems as potential candidates for BACE inhibition.
Researchers at Seeon were intrigued by the opposite effects the BACE inhibitor and immunotherapy appear to have on hyperactivity. Some asked if the type of immunotherapy used might be critical, since some antibodies might solubilize toxic forms of Aβ, while others corral them. “From a therapeutic perspective, this is extremely important question to answer,” agreed Busche.
At a more basic level, researchers still have no idea what kick-starts the dystrophy that might be a driving force behind many of these electrical disturbances in the brain. Hints came from Giuseppina Tesco, who studies BACE1 trafficking in her lab at Tufts University, Boston. In prior work, she found that transport of the protease depends on the GGA3 protein, which helps recycle other proteins from the cell membrane (see Jun 2007 news; Dec 2013 conference news). Most of the work on GGA3 and BACE has been done in non-polarized cells, but now Selene Lomoio in Tesco’s lab did this in primary neurons.
Lomoio capitalizes on the super-high resolution of total internal reflection fluorescence microscopy to track the movement of molecules in living cells. Tesco showed live images of both GGA3 and BACE1 scooting along axons (see movie below). Both travelled antero- and retrogradely at about the same speed, though BACE sped along faster than GGA3, suggesting that other carriers might contribute to trafficking of the protease. Nonetheless, in GGA3 knockout neurons, most BACE vesicles just stopped, even though other proteins, such as synaptophysin, motored along at normal speed. Furthermore, in GGA3 KOs, BACE accumulated in swollen axons that looked very like dystrophic neurites. Tesco said the swellings most likely reflect defective trafficking of molecules to the lysosome for degradation.
Others were impressed by the quality of Tesco’s data and peppered her with questions about what it might imply about dystrophic neurites. Tesco does not know if GGA3 levels drop in dystrophic neurites in AD—unfortunately, GGA3 antibodies are not good enough for immunostaining. Others wondered if any toxic Aβ species block GGA trafficking or if loss of GGA3 leads to an increase in production of Aβ from APP. Tesco agreed all these were testable hypotheses. “This experimental approach could yield mechanistic insight into the accumulation of BACE1 in dystrophic neurites in AD,” she said.
Tesco’s findings gibe with work from Vassar’s lab at Northwestern University, Chicago. He reported that BACE1 accumulates in dystrophic neurites in AD, where it increases cleavage of APP and releases Aβ (see Sadleir et al., 2016).—Tom Fagan
While BACE inhibitor trials are unfolding, more than 100 experts in the field gathered for the 2nd Kloster Seeon Meeting on BACE Proteases in Health and Disease, held September 25-27 near Munich. Researchers are encouraged that no major side effects have yet reared their ugly heads, but even so, safety worries still inspire active research toward a new round of drugs. In Seeon, researchers described new assays to spot off-target effects, new insight into why fading hair color is the most common problem seen in BACE knockout mice, and new inhibitors that selectively target BACE1 over BACE2. “I think we are on the verge of seeing the next generation of BACE inhibitors,” said Robert Vassar of Northwestern University, Chicago, who co-organized the meeting. “I was not sure this was possible, but now it seems to be happening.”
With BACE, a Picky Drug is a Good Thing
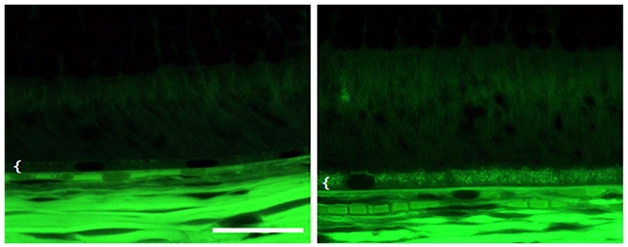
Like any other drug, BACE inhibitors can have both on-target and off-target side effects. Scientists believe the latter led to the eye toxicity that stopped clinical trials of the first BACE inhibitors (see Mar 2011 conference news). More specifically, the assumption has been that by also blocking cathepsin D, another aspartyl protease, BACE1 inhibitors prevented the normal turnover of photoreceptors in the eye, causing lipofuscinosis. In Seeon, Douglas Johnson from Pfizer, Cambridge, Massachusetts, put any lingering doubt about this scenario to rest.
Eye Tox. Autofluorescenece microscopy of rat retina shows the BACE inhibitor PF-9283 (right) disrupts the retinal pigment epithelium (white bracket). [Image courtesy of Douglas Johnson, Nature Communications.]
Johnson used photoaffinity versions of BACE inhibitors to covalently label whatever they bind to in retinal epithelial cells. Then, in an unbiased fashion, he identified those targets by mass spectrometry (see Zuhl et al., 2016). He found that PF-9283, a BACE inhibitor Pfizer developed, showed high affinity for cathepsin D. In competition experiments, LY2811376, one of Eli Lilly’s early BACE inhibitors, and CP-108671, a renin protease inhibitor, both prevented PF-9283 from binding cathepsin D, showing that these compounds also had high off-target affinity for the protease. In contrast, LY2886721 did not, confirming reports that this compound is much more selective for BACE than other aspartyl proteases.
That confirmation aside, Johnson cautioned that test-tube assays for cathepsin D inhibition should be taken with a grain of salt. He found that the affinity of PF-9283 for cathepsin D was two orders of magnitude higher in cells than in vitro. Stefan Lichtenthaler, German Center for Neurodegenerative Diseases in Munich, thought this was astounding. Furthermore, in an exposure-response analysis, in which Johnson correlated IC50s of various compounds with eye epithelium damage in rats, only the cellular IC50s predicted toxicity. “These are very important revelations for drug development,” said Lichtenthaler. “This suggests that scientists who design new inhibitors need to run cell-based assays to determine selectivity,” he told Alzforum. While it remains to be proven why some inhibitors have higher affinity for cathepsin D in cells, it may come down to the compound’s accumulation in acidic environments, since this protease works mostly in the lysosome.
Eye toxicity aside, scientists continue to worry about on-target side effects as well. Importantly, all BACE inhibitors currently in trials block both BACE1 and BACE2. While the latter seems to have fewer substrates and is less active in the brain than BACE1, it may still underlie some unwanted effects, such as the fur discoloration seen in mice. Scientists are also concerned that long-term use of inhibitors might bring to light new BACE2-related effects. Could they develop BACE1-selective compounds to take a cleaner shot at the target?
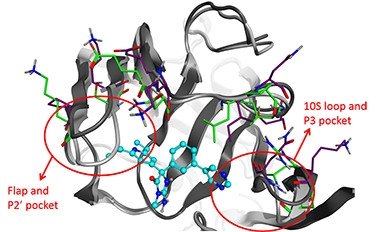
BACE Details.
Overlapping X-ray crystallographic structures of BACE1 (dark gray, blue side chains) and BACE2 (light gray, green side chains) reveal differences (red circles) at the active site that researchers might exploit to develop BACE1-specific inhibitors (turquoise and blue). [Image courtesy of Dieder Moechars.]
Dieder Moechars and colleagues at Janssen Research and Development, Beerse, Belgium, have done just that. Moechars’ team took advantage of the known crystal structures of BACE1 and 2 to design drugs that bind the former but not so much the latter. The researchers noted that because BACE2 has a more rigid active site, it might restrict compounds that the BACE1 cleft would allow in (see image at left). He illustrated this by comparing co-crystal structures of BACE1 and BACE2 together with an inhibitor developed by Wyeth that has about 500-fold higher affinity for BACE1 (see Malamas et al., 2010). Janssen researchers have now improved substantially on that, developing compounds topping out at 7,000-fold higher affinity for BACE1. Janssen plans to select one of these for clinical development early next year, Moechars said.
“This will help allay concerns about hypopigmentation or blocking cleavage of BACE2 substrates in the pancreas,” said Vassar. “Even though BACE2 knockouts are fairly normal, we need to be concerned about blocking BACE2,” he said. Vassar also noted that these new compounds are more potent inhibitors of BACE1, which would translate to lower doses in the clinic. “That means much lower potential for off-target side effects,” he said. “These compounds seem to have lots of advantages.”
Rabbit of a Different Color. Dutch belted rabbits treated for four months with verubecestat (right) lose pigment in their coats. [Image courtesy of Lynn Hyde, Matthew Kennedy, Merck Research Labs.]
What about selectivity against BACE1 substrates other than APP? That idea may not be so far-fetched. After all, γ-secretase modulators can shift cleavage of the APP C-terminal fragment (CTF) while allowing the secretase to process Notch, though these compounds, too, have run into toxicity problems at the preclinical stage. Moechars said Janssen scientists are searching for an APP-selective inhibitor. They want a compound that binds to APP and prevents it from coming into contact with BACE1. With this in mind, they devised an assay for compounds that block production of CTF from APP even while allowing processing of neuregulin1. The researchers had to screen 1.2 million compounds to turn up one that fit the bill. “It proves the concept, but we have to use micromolar amounts, making it far from an ideal inhibitor,” said Moechars. Researchers were intrigued, but wondered how efficient this approach would be in practice. Since the substrate has to be blocked, not the catalyst, some questioned whether enough drug can be delivered into the brain to do the job.
As for the inhibitors currently in trials, most of them seem to mimic the loss of coat color seen in BACE knockouts (see image above). How might that manifest in people? If scientists knew, no one at Seeon was willing to say out loud. Lynn Hyde from Merck, Kenilworth, New Jersey, noted that reports of skin or hair color change in BACE inhibitor trials are scant. Since the trials are still blinded, no firm conclusions can be drawn, though on the face of it such effects would seem to be pretty obvious to participants and trial staff.
To begin to probe this question, Hyde has looked for underlying causes of color loss in mice. She examined changes in gene expression in the skin during anagen phase, the period during which hair grows from follicles and becomes pigmented. Hyde compared gene expression in control animals with that in mice given the BACE inhibitor MBI-3. The drug suppressed expression of 18 genes, including BACE2 and proteins known to be involved in pigmentation such as PMEL17 and tyrosinase. In the BACE1/2 double knockouts generated at Merck, Hyde found the same 18 genes to be suppressed. All of them lie downstream of microphthalmia-associated transcription factor, a major regulator of pigmentation. Hyde said this gene expression signature might serve as a biomarker correlating with BACE inhibitor effects on pigmentation. She does not know if BACE processes MiTF, but said that would be worth exploring.
Tracking BACE Inhibition
One way to mitigate side effects of BACE inhibition would be to find a dose range that suppresses Aβ accumulation while allowing sufficient processing of other substrates. Lichtenthaler reported on a proteomics approach to monitor substrate cleavage by measuring levels of their shed ectodomains in the CSF. Researchers at the meeting thought this might be a good way to test if cleavage of substrates other than APP is being spared (see Part 3 of this series).
For APP itself, researchers have long used Aβ levels in the CSF to gauge efficiency of BACE inhibition; however, Mathias Jucker, University of Tubingen, Germany, reported that this data may be difficult to interpret (see Schelle et al., 2016). APP/PS1 mice, for example, accumulate robust amyloid plaques in the brain by 7.5 months of age, while their CSF Aβ falls about threefold. In contrast, when 1.5-month-old animals ate food pellets laced with the BACE inhibitor NB-360, they accumulated no plaques in the brain over the next six months, but their CSF Aβ fell just the same. Because both plaque formation and BACE inhibition reduce the level of CSF Aβ, the latter may be a misleading marker of BACE activity, said Jucker. This holds in a treatment paradigm as well, he said. If begun when APP23 mice are 15 months old and have rampant plaques, 6.5 months’ worth of treatment with NB-360 reduced CSF Aβ levels, but CSF Aβ also fell in untreated animals.
Jucker proposed a different marker to track BACE processing of APP, and it is none other than tau. Previously, Stephan Käser in Jucker's lab reported that tau climbs in the CSF of transgenice APP/PS mouse models of AD as Aβ plaques begin to accumulate. This happens even though the animals show no tau pathology or neuronal loss (see Jul 2013 news). Would CSF tau respond to BACE inhibition?
Jucker and colleagues developed a highly sensitive, single-molecule array immunoassay (Simoa) for tau that can measure the protein in CSF of young, normal mice and in transgenic animals before they develop overt pathology. He used it to test the effect of NB-360. At Seeon, Jucker reported that in both the APPPS1 prevention paradigm and in the APP23 treatment case, the BACE inhibitor lowered CSF tau while CSF tau climbed in untreated animals. “CSF tau could be a very valuable marker to predict the efficacy of BACE inhibitors in clinical trials,” Jucker said.
He further hinted that another up-and-coming marker of neurodegeneration, neurofilament light chain (NfL), responds to BACE inhibitors, as well. That could be even more valuable, since Jucker has reported that blood NfL tracks disease progression (see Jun 2016 news).
While researches wondered why tau and NfL are released into the CSF in the absence of overt neurodegeneration, they were cautiously optimistic about these data, saying they represent a real breakthrough. “We don’t have a blood biomarker for any neurodegenerative disease yet,” noted Vassar, adding, “These data are extremely interesting and potentially very important for the field.”—Tom Fagan





Comments
No Available Comments
Make a Comment
To make a comment you must login or register.