Researchers have tied sleep to clearance of waste products such as excess Aβ. New research suggests this clearance may be driven by a change in the extracellular ion composition, which swells the interstitial fluid. Other work finds an essential role for a signaling pathway involving the kinase complex mTORC1 and the electrical rhythms of REM sleep. Read Madolyn Rogers' two-part series.
Series
To Sleep, Perchance to Clean…the Brain, Make Memories
Sleep and Brain Cleansing—Fresh Insights into Regulation and Disruption
Throughout the arc of human history, sleep has been a mysterious process that has captured the imaginations of both artists and scientists. Researchers are slowly deciphering how sleep restores us, and how its woeful absence makes us ill. Scientists now think a good night’s rest benefits the brain by clearing away cellular waste and laying down memories. Recent studies on how these processes work may have implications for neurodegenerative disease, particularly Alzheimer’s. “There is an increasing awareness that we need to know more about how sleep is implicated in disease etiologies,” said Maiken Nedergaard at the University of Rochester Medical Center, New York, who led one of the studies.
This story explains how sleep and osmotic pressure are intimately connected, how poor fluid flow in the brain correlates with disease, and how brain injury may disturb sleep. (For new insights into the role of sleep in memory, see Part 2 of this series.)
Sleeping by Osmosis
Both the volume and flow of extracellular fluid are known to surge when the brain catches some zzz's. This extra flow facilitates the clearance of solutes including Aβ, according to one theory.
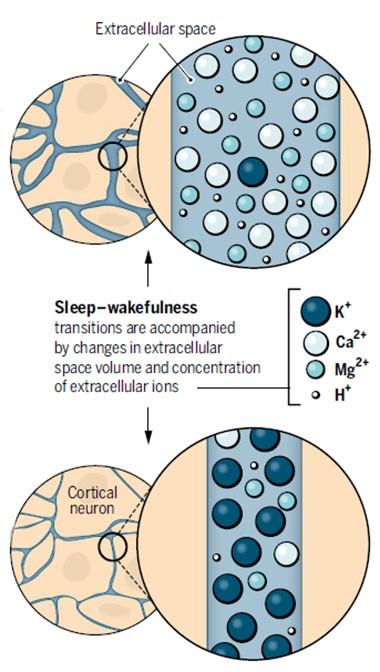
Night and Day.
During sleep (top), calcium and magnesium flood into the extracellular fluid and its volume swells, while in the waking brain, potassium predominates and volume shrinks (bottom). [Courtesy of Science/AAAS.]
Nedergaard wanted to know why there is more fluid in sleeping brains. Because osmotic forces control fluid movements, Nedergaard speculated that the answer might have something to do with the extracellular concentrations of ions and solutes. As reported in the April 29 Science, joint first authors Fengfei Ding and John O’Donnell and their colleagues tested this idea by adding a “wakeup” mix of neuromodulators to mouse cortical brain slices. The mix included norepinephrine, acetylcholine, and dopamine, which are produced by brainstem and basal brain regions and are known to stimulate waking patterns of electrical activity in the brain. The treatment induced a rapid and persistent rise in extracellular potassium in the slices. Neurons release this ion when they fire, but surprisingly, potassium spiked even when brain activity in the slices was silenced with tetrodotoxin. This demonstrated that the ion change occurred independently of electrical activity. It is not yet clear what caused the potassium efflux, Nedergaard said.
Intrigued, the authors measured ion concentrations in waking and sleeping mice via microdialysis. As in brain slices, potassium levels shot up within a few seconds of waking; they remained high until the animals nodded off, then plunged. These changes occurred even when electrical activity was dampened with an AMPA receptor antagonist. Meanwhile, calcium and magnesium ions waxed and waned in opposition to the potassium, rising during sleep and falling as animals awoke. Their transitions were a bit slower, taking about a minute apiece. The ion changes all occurred independently of each other, the scientists reported.
To tease out the effect of different ion concentrations, the authors infused ion cocktails into the brains of mice through a cranial window. When they added a “waking” mix of ions to one cortical region of a sleeping mouse, the neurons in that small region shifted into waking patterns of electrical activity, although the mouse remained asleep. Likewise, a “sleeping” mix added to awake animals induced slow-wave electrical activity characteristic of sleep in that teeny portion of cortex, even while the animals remained active. At the same time, the volume of local extracellular fluid shrank or expanded in keeping with the ion mix (see image above). This was determined by the classic method of infusing the cation tetramethylammonium and recording how quickly it diffused from the delivery site.
Then the authors went one step further. They changed the ion composition throughout the brain by infusing ion cocktails into the cerebrospinal fluid. With that, they found they could transition global brain activity, waking up sleeping mice and putting awake mice to sleep.
The findings suggest, at a simple level, that ion concentration itself controls sleep/wake in the brain, Nedergaard told Alzforum. The idea is not new; studies from the 1930s first proposed that calcium and magnesium promoted sleep, while potassium promoted wakefulness, noted Hans-Peter Landolt and Sebastian Holst at the University of Zurich, in an accompanying editorial. However, this idea fell into obscurity when subsequent research assumed that changes in electrical activity caused the ups and downs in ion concentrations during sleep and wakefulness. The new data imply the opposite, Nedergaard noted. As an animal falls asleep, extracellular potassium plummets first, hyperpolarizing and silencing neurons at the same time fluid volume swells to facilitate clearance of solutes. “When the brain needs to clean itself, it shuts itself off by lowering potassium,” Nedergaard suggested.
Brendan Lucey at Washington University in St. Louis found the data surprising. “I wouldn’t have thought that regulating ion concentrations in mouse CSF would result in state changes in the brain,” he told Alzforum. He noted that the study used careful controls and built its case well, moving from brain slices to live animals.
What Can Go Wrong
Nedergaard thinks the research may also provide clues to what goes awry in disease states. A number of studies have linked disrupted sleep to Alzheimer’s risk (see, e.g., Aug 2012 conference news; Oct 2013 news). Recent work indicates that Aβ exits the brain primarily during sleep, suggesting one possible reason for the association (see Sep 2009 news; Apr 2013 conference news; Jun 2014 news). Nedergaard and colleagues had previously proposed a mechanism for this clearance. They described a “glymphatic” flow that washes through the brain interstitial space, dragging Aβ and other solutes along with it. Specialized channels on astrocytes that pump water power the flow, which is much greater during sleep (see Aug 2012 news; May 2014 conference news). The new data suggest the increase in osmotic pressure that occurs during sleep may help power that current. “The data help establish a mechanism for Nedergaard’s prior observations that Aβ clearance occurs during sleep,” Lucey said.
Ion imbalance might underlie other symptoms as well. For example, Ding and colleagues found that in animals recovering from anesthesia, the ions lagged in returning to normal waking levels, with potassium taking a couple of minutes and calcium and magnesium requiring up to half an hour to stabilize. Mice in this state appeared disoriented, running into the walls of their cage. This slow transition may explain the grogginess common upon waking from anesthesia, Nedergaard speculated. Perhaps a similar ion perturbation causes some of the confusion seen in people with Alzheimer’s, particularly in the evenings, Nedergaard said. In ongoing work, she is measuring ion concentrations in the brains of aged AD model mice to test this idea. She is also interested in whether extracellular potassium levels continue to climb if animals are kept awake past their normal hour for shut-eye. High potassium is known to cause seizures, and might provide an explanation for why these occur after prolonged sleep deprivation, Nedergaard noted.
The findings might also point toward therapeutic targets, Landolt and Holst suggested. “Pumps and transporters that control ion flow across cell membranes may be promising new targets for treating sleep-wake disorders,” they wrote. For her part, Nedergaard noted that astrocytes express specific subtypes of sodium/potassium pumps, suggesting these could be selectively targeted.
Beyond Glymph—Sleep and Other Clearance Pathways
Glymphatic flow is but one of several complementary routes for clearing solutes from the brain. Researchers last year reported the discovery of lymphatic vessels in the dura mater that absorb fluid from interstitial fluid (ISF) and CSF and transport it to cervical lymph nodes (see Louveau et al., 2015; Aspelund et al., 2015). In addition, fluids can drain along the basement membranes of brain blood vessels, through the perivascular spaces. Enlargements in these Virchow-Robin spaces, as they are called, correlate with cardiovascular disease, stroke, cognitive problems, and dementia.
Researchers led by Joel Ramirez at the University of Toronto have associated low sleep quality with anatomical changes in these fluid-filled gaps. Along with Courtney Berezuk, Ramirez examined data from 26 adults with an average age of 60 who were evaluated for cardiovascular disease (see Berezuk et al., 2015). Participants with the largest Virchow-Robin spaces slept worst. They took longer to fall asleep, woke up more during the night, and spent less time in the third and deepest stage of non-REM sleep.
Why these spaces were enlarged remains unclear. The authors speculated that lack of sleep rendered drainage from perivascular spaces inefficient, which in turn could cause these channels to clog up with waste products such as Aβ and expand (see Weller et al., 2009). On the other hand, underlying cardiovascular disease by itself might be responsible for these spaces ballooning, regardless of sleep problems. One theory holds that as arteries harden, they beat more against surrounding glia and carve out a larger space, said Costantino Iadecola at Weill Cornell Medical Center, New York. The consequences of enlarged Virchow-Robin spaces are unknown. Berislav Zlokovic at the University of Southern California, Los Angeles, pointed out that it might influence the rate of solute clearance from the ISF to the CSF. However, Iadecola noted that remains to be proven (see full comment below).
Other studies support the idea that normal clearance mechanisms falter in AD. Researchers led by Tsutomu Nakada at the University of Niigata, Japan, reported that the flow of water from ISF into CSF slows down in older people, and lags even more in those with Alzheimer’s disease (see Suzuki et al., 2015). They compared fluid flow in 10 young volunteers, 10 healthy aged volunteers, and 10 people diagnosed with AD dementia, using PET imaging of water labeled with an oxygen isotope. The flow in AD patients was about half that in the younger people, with healthy elderly falling about halfway between the two groups.
John Cirrito at WashU noted that these findings fit with previous evidence from his WashU colleague Randy Bateman, who found that rather than spiking during sleep as they do in healthy adults, CSF Aβ levels remain steady throughout the circadian cycle in AD patients (see Dec 2010 news; Aug 2012 conference news; Huang et al., 2012). “Nakada’s study suggests the reason for flat Aβ levels is because the flow from ISF to CSF is impaired,” Cirrito said.
Beyond AD
Perturbed sleep shows up in other brain disorders as well. In the April 27 Neurology, researchers led by Lukas Imbach and Christian Baumann at University Hospital Zurich, reported that people with traumatic brain injury sleep more than controls even 18 months after their injury. The findings extended data from a previous study, where the authors reported increased sleepiness in a cohort of 42 young adults six months after TBI (see Imbach et al., 2015). At 18 months, the authors were able to obtain additional polysomnography data from 31 of those participants. Compared with 42 age-matched controls, the TBI patients slept about one hour more per night, but nonetheless remained sleepier during the day, as measured by how quickly they fell asleep for naps. Overall, two-thirds of the TBI patients were diagnosed with excessive daytime sleepiness, as compared to one-fifth of controls. Other sleep parameters appeared normal. The findings match the data at six months, indicating the changes in sleep patterns persisted.
Unlike at six months after their injury, at 18 months no correlation emerged between sleep problems and the severity of the injury. It may be that the need for extra sleep soon after injury serves a purpose in healing the brain, but over time sleep disturbances transform into a chronic, maladaptive condition, the authors speculated. Intriguingly, a recent study from the same group found that when rats were induced to sleep more in the first two weeks after a traumatic brain injury, they maintained their ability to remember new objects, whereas controls did not. These power sleepers expressed less APP in the hippocampus and cortex than injured controls did, suggesting the extra sleep might mitigate axonal injury (see Morawska et al., 2016). However, it remains to be determined whether the extra sleep TBI patients get more than a year after injury helps or harms their brains.
Notably, these patients did not realize they had a sleep problem, as their self-reports and clinical interviews regarding sleep habits resembled those of controls. This suggests sleep disorders may go unrecognized in TBI patients, and therefore untreated. Such patients should be evaluated by objective sleep laboratory measures, the authors suggest.
“Imbach et al. make a compelling case that post-traumatic sleep-wake disorders may represent a silent epidemic,” wrote Brian Edlow at Massachusetts General Hospital, Boston, and Gert-Jan Lammers at Leiden University Medical Centre, the Netherlands, in an accompanying editorial. They noted that the study represents the most comprehensive longitudinal analysis of post-traumatic sleep-wake disturbances to date.
Would sleep therapy help TBI patients, or people with AD for that matter? No one knows. “We need very good controlled, prospective studies on how sleep therapy (not sleep medications) can reduce the progression of different diseases, including Alzheimer’s,” Nedergaard said.—Madolyn Bowman Rogers
References
News Citations
- In mTORC and Theta Rhythms, New Clues to How Sleep Locks Down Memories
- Night Owl? Early Bird? Good Night’s Sleep May Protect the Brain
- From ApoE to Zzz’s—Does Sleep Quality Affect Dementia Risk?
- Sleep Deprivation Taxes Neurons, Racks Up Brain Aβ?
- Sleep Patterns, Circadian Clock Linked to Aβ Oxidative Stress
- While You Were Sleeping—Synapses Forged, Amyloid Purged
- Brain Drain—“Glymphatic” Pathway Clears Aβ, Requires Water Channel
- Glymphatic Flow, Sleep, microRNA Are Frontiers in Alzheimer’s Research
- Paper Alert: In Vivo Human Data Shows Reduced Aβ Clearance in AD
- Plaque May Quash Seesawing CSF Aβ Levels
Paper Citations
- Louveau A, Smirnov I, Keyes TJ, Eccles JD, Rouhani SJ, Peske JD, Derecki NC, Castle D, Mandell JW, Lee KS, Harris TH, Kipnis J. Structural and functional features of central nervous system lymphatic vessels. Nature. 2015 Jul 16;523(7560):337-41. Epub 2015 Jun 1 PubMed.
- Aspelund A, Antila S, Proulx ST, Karlsen TV, Karaman S, Detmar M, Wiig H, Alitalo K. A dural lymphatic vascular system that drains brain interstitial fluid and macromolecules. J Exp Med. 2015 Jun 29;212(7):991-9. Epub 2015 Jun 15 PubMed.
- Berezuk C, Ramirez J, Gao F, Scott CJ, Huroy M, Swartz RH, Murray BJ, Black SE, Boulos MI. Virchow-Robin Spaces: Correlations with Polysomnography-Derived Sleep Parameters. Sleep. 2015 Jun 1;38(6):853-8. PubMed.
- Weller RO, Boche D, Nicoll JA. Microvasculature changes and cerebral amyloid angiopathy in Alzheimer's disease and their potential impact on therapy. Acta Neuropathol. 2009 Jul;118(1):87-102. PubMed.
- Suzuki Y, Nakamura Y, Yamada K, Igarashi H, Kasuga K, Yokoyama Y, Ikeuchi T, Nishizawa M, Kwee IL, Nakada T. Reduced CSF Water Influx in Alzheimer's Disease Supporting the β-Amyloid Clearance Hypothesis. PLoS One. 2015;10(5):e0123708. Epub 2015 May 6 PubMed.
- Huang Y, Potter R, Sigurdson W, Santacruz A, Shih S, Ju YE, Kasten T, Morris JC, Mintun M, Duntley S, Bateman RJ. Effects of age and amyloid deposition on aβ dynamics in the human central nervous system. Arch Neurol. 2012 Jan;69(1):51-8. PubMed.
- Imbach LL, Valko PO, Li T, Maric A, Symeonidou ER, Stover JF, Bassetti CL, Mica L, Werth E, Baumann CR. Increased sleep need and daytime sleepiness 6 months after traumatic brain injury: a prospective controlled clinical trial. Brain. 2015 Mar;138(Pt 3):726-35. Epub 2015 Jan 15 PubMed.
- Morawska MM, Büchele F, Moreira CG, Imbach LL, Noain D, Baumann CR. Sleep Modulation Alleviates Axonal Damage and Cognitive Decline after Rodent Traumatic Brain Injury. J Neurosci. 2016 Mar 23;36(12):3422-9. PubMed.
Further Reading
In mTORC and Theta Rhythms, New Clues to How Sleep Locks Down Memories
Parents often insist their children get a good night’s sleep before a big exam, and science says they are onto something. Studies have demonstrated that without sleep, memory traces from the day before vanish. Exactly how sleep strengthens memories, however, remains fuzzy. New studies offer cellular and molecular insights. One reports a protein synthesis pathway essential for memory consolidation, while the second details how consolidation depends on the coordinated firing of neurons in the hippocampus during rapid-eye-movement sleep. Such theta rhythms can falter in people with Alzheimer’s disease. Together the findings offer scientists clues to what might go wrong in people with sleep problems, and may suggest ways to compensate. For more on how sleep helps the brain rid itself of unwanted protein, see Part 1 of this story.
TORC Drives Memories
In the April 26 Science Signaling, researchers led by Ted Abel at the University of Pennsylvania, Philadelphia, pinpointed a complex, mTORC. This kinase complex activates protein translation and is essential for memory consolidation during sleep, the scientists report. Keeping mice awake dampened mTORC activity and the mice flubbed a memory test, whereas artificially stimulating mTORC1 signaling maintained memories even in sleep-deprived animals. “This is the first study to functionally manipulate protein synthesis and reverse the effects of sleep deprivation,” Abel told Alzforum. The findings highlight a crucial role for protein production in memory formation during sleep, he added.

Connecting Sleep and Memory.
Sleep-deprived mice had less mTORC1 signaling, leading to less protein synthesis and a failure to consolidate memories. [Courtesy of Science Signaling/AAAS.]
Commenters called the study a step forward. “The authors have placed our understanding of the molecular neurobiology of sleep deprivation and its deleterious effects on memory cognition on a firm foundation,” wrote David Sweatt and Kimberly Hawkins in an accompanying editorial. Alcino Silva at the University of California, Los Angeles, wrote to Alzforum, “To my knowledge this is the first time that anyone has found a specific molecular pathway, and even better, a specific molecular manipulation, that may help us address the sleep deprivation epidemic that plagues the modern world.” To find such a specific mechanism was surprising, and opens the door to possible interventions to dampen the effects of sleep deprivation, he added. Such interventions might also be applicable to Alzheimer’s and other neurodegenerative diseases in which sleep is often disrupted.
Researchers have long wondered why lack of sleep degrades memory (for review, see Maquet, 2001; Abel et al., 2013). Most research in this area has focused on neurotransmitters and changes in neuronal firing patterns during sleep, but a handful of papers linked protein synthesis during slumber to memory (see Ramm and Smith, 1990; Nakanishi et al., 1997; Seibt et al., 2012). Previously, Abel and colleagues compared hippocampal gene expression in sleep-deprived mice to that in well-rested littermates. They found a marked drop in proteins involved in translation, particularly the mammalian target of rapamycin (mTOR). This cytoplasmic kinase forms part of the insulin signaling pathway and stimulates protein synthesis, although it also subserves cell survival and proliferation (see Vecsey et al., 2012).
Because protein synthesis has been linked to synaptic plasticity, the authors investigated whether it was the process connecting memory impairment to sleep deprivation (see Bourtchouladze et al., 1998; Kelleher et al., 2004; Abraham et al., 2008). First author Jennifer Tudor compared the production of new proteins in the hippocampi of mice that had been deprived of sleep for five hours to that in mice who had just awakened from five hours of slumber. She found a dramatic difference: The sleep-deprived animals had made about half as many new proteins during those five hours as the sleeping ones.
The authors next examined proteins known to interact with mTOR for changes in sleep-deprived mice. They found that lack of sleep kept mTOR from binding Raptor, aka regulatory-associated protein of mTOR, to form the mTORC1 complex. This complex has been studied in depth, and scientists have worked out how it acts in a series of regulated steps that lead to activation of protein synthesis. First, mTORC1 phosphorylates eukaryotic translation initiation factor 4E-binding protein 2 (4EBP2). Phosphorylation of 4EBP2 prevents it from binding to and sequestering eukaryotic initiation factor 4E (eIF4E), and thus frees eIF4E to bind to the related protein eIF4G and turn on translation at the ribosome. In effect, then, sleep releases the brakes on protein translation by activating mTORC1 to phosphorylate 4EBP2 (see image below). Sleep-deprived mice, which did not form the mTORC1 complex, made about half as much phosphorylated 4EBP2 as their rested kin. After they were allowed to sleep, phosphorylated 4EBP2 rose again. Sleep deprivation did not change other proteins known to be involved in this protein synthesis pathway, including the kinase S6K that activates ribosomes, suggesting mTORC1 and 4EBP2 were specifically affected.
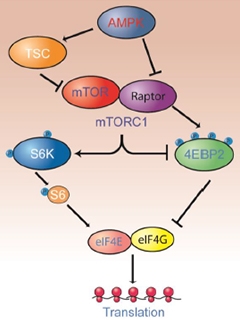
Sleep Toggles Translational Switch.
During sleep, mTORC1 activity inhibits 4EBP2 and activates S6 kinase, releasing the brakes on protein translation in neurons. [Courtesy of Science Signaling/AAAS.]
To find out if this mechanism truly contributed to memory formation during sleep, the authors overexpressed 4EBP2 in hippocampal excitatory neurons using a viral vector. In theory, this overexpression should bump up levels of the phosphorylated version. Three weeks later, mice that received the vector indeed had four times as much phosphorylated 4EBP2 in their hippocampi as did controls—even after sleep deprivation. As predicted, the strategy protected their memories from the effects of lost sleep. The sleepy animals maintained normal amounts of eIF4E bound to eIF4G, and made as much new protein as did rested controls. Moreover, they remembered the locations of objects they had seen before as well as did the rested mice. Sleep-deprived mice injected with a control vector, on the other hand, make less new protein and botched the memory test.
In future work, Abel plans to dissect the regulation of 4EBP2 by looking at specific phosphorylated residues and which phosphatases affect them. He also wants to identify upstream and downstream components of the pathway, by investigating what kicks off signaling through mTORC1 and what proteins are made as a result. In awake animals, insulin triggers this protein-synthesis pathway. Intriguingly, sleep deprivation can lead animals (and people) to eat more, Abel noted. The sugar in food elicits insulin release, thus dialing up protein synthesis and perhaps making up for the lack of it after sleep deprivation. “It would be interesting to see if eating is a compensatory mechanism [for lost sleep],” Abel suggested. In addition, because the same type of memories that are impaired by sleep deprivation also falter in cognitive impairment and dementia, he wondered if similar mechanisms might be at work there.
Sleep Rhythms and Memory
Sleep is not a uniform process. Rather, its stages are distinguished by varying neuronal activity. Are some of these stages more important for memory than others? Researchers led by Sylvain Williams at McGill University, Montreal, and Antoine Adamantidis at the University of Bern, Switzerland, addressed this question in the May 13 Science. They homed in on electrical activity during REM sleep. Low-frequency (i.e., 4-7 Hz) theta rhythms in the brain distinguish REM from other sleep stages, and hippocampal theta rhythms have been linked to memory consolidation (see Poe et al., 2000; Louie and Wilson, 2001). Some studies have correlated disruptions in REM with faulty memory formation, but because REM is difficult to manipulate, researchers could not prove a causal connection (for review, see Stickgold and Walker, 2005). To address this, the authors turned to optogenetics, in which neural circuits are switched on and off with flashes of light (see Nov 2012 series).
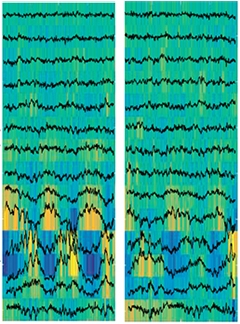
Quieting Theta Rhythms.
Optogenetically silencing GABAergic neurons in the medial septum dampened (right) the theta waves seen normally (left) in the sleeping hippocampus. [Courtesy of Science/AAAS.]
First author Richard Boyce injected a viral vector containing either a photosensitive ion channel or just a fluorescent tag into the medial septum of the brains of transgenic mice. The mice also expressed an activator for the vector, but only in GABAergic neurons of the medial septum. These inhibitory neurons project to the hippocampus and are believed to pace theta rhythms there. The authors then delivered a pulse of light to the medial septum, hyperpolarizing and silencing the neurons. When delivered during REM sleep, this light treatment dropped theta power in the hippocampus by two-thirds. Other parameters of sleep remained normal.
The authors allowed transfected and control mice to explore a cage containing two new objects before the animals bedded down for four hours of sleep. During sleep, researchers optogenetically disrupted REMs in some of the animals. Afterward, they re-introduced them to the two objects, one of which was in a new location. Mice with disrupted REM sleep explored both objects equally, suggesting they did not remember either. Control mice, as well as those that did not receive light stimulation, or only received it during non-REM sleep, remembered their previous play session and mostly investigated the altered toy. The authors obtained similar results using a contextual fear conditioning test: Mice that lacked normal REM sleep failed to remember previous training.
Both of these cognitive tests involve spatial memory. The data suggest a role for REM sleep theta rhythms in locking down hippocampal spatial memories in particular, the authors noted. Intriguingly, some evidence indicates that Alzheimer’s patients have disrupted theta rhythms (see Aug 2010 news). People with AD also have trouble with spatial navigation, frequently becoming lost in their own neighborhoods or even houses (see Mar 2007 news; Oct 2015 news). Whether poor sleep plays a role in this deficit remains to be determined.—Madolyn Bowman Rogers
References
News Citations
- Sleep and Brain Cleansing—Fresh Insights into Regulation and Disruption
- Off Key—Aβ Detunes the Theta Rhythms of the Hippocampus
- “Morris Land Maze” Reveals Early Loss of Spatial Memory in MCI
- Young ApoE4 Carriers Wander Off the ‘Grid’ — Early Predictor of Alzheimer’s?
Series Citations
Paper Citations
- Maquet P. The role of sleep in learning and memory. Science. 2001 Nov 2;294(5544):1048-52. PubMed.
- Abel T, Havekes R, Saletin JM, Walker MP. Sleep, plasticity and memory from molecules to whole-brain networks. Curr Biol. 2013 Sep 9;23(17):R774-88. PubMed.
- Ramm P, Smith CT. Rates of cerebral protein synthesis are linked to slow wave sleep in the rat. Physiol Behav. 1990 Nov;48(5):749-53. PubMed.
- Nakanishi H, Sun Y, Nakamura RK, Mori K, Ito M, Suda S, Namba H, Storch FI, Dang TP, Mendelson W, Mishkin M, Kennedy C, Gillin JC, Smith CB, Sokoloff L. Positive correlations between cerebral protein synthesis rates and deep sleep in Macaca mulatta. Eur J Neurosci. 1997 Feb;9(2):271-9. PubMed.
- Seibt J, Dumoulin MC, Aton SJ, Coleman T, Watson A, Naidoo N, Frank MG. Protein synthesis during sleep consolidates cortical plasticity in vivo. Curr Biol. 2012 Apr 24;22(8):676-82. Epub 2012 Mar 1 PubMed.
- Vecsey CG, Peixoto L, Choi JH, Wimmer M, Jaganath D, Hernandez PJ, Blackwell J, Meda K, Park AJ, Hannenhalli S, Abel T. Genomic analysis of sleep deprivation reveals translational regulation in the hippocampus. Physiol Genomics. 2012 Oct 17;44(20):981-91. Epub 2012 Aug 28 PubMed.
- Bourtchouladze R, Abel T, Berman N, Gordon R, Lapidus K, Kandel ER. Different training procedures recruit either one or two critical periods for contextual memory consolidation, each of which requires protein synthesis and PKA. Learn Mem. 1998 Sep-Oct;5(4-5):365-74. PubMed.
- Kelleher RJ 3rd, Govindarajan A, Tonegawa S. Translational regulatory mechanisms in persistent forms of synaptic plasticity. Neuron. 2004 Sep 30;44(1):59-73. PubMed.
- Abraham WC, Williams JM. LTP maintenance and its protein synthesis-dependence. Neurobiol Learn Mem. 2008 Mar;89(3):260-8. Epub 2007 Nov 7 PubMed.
- Poe GR, Nitz DA, McNaughton BL, Barnes CA. Experience-dependent phase-reversal of hippocampal neuron firing during REM sleep. Brain Res. 2000 Feb 7;855(1):176-80. PubMed.
- Louie K, Wilson MA. Temporally structured replay of awake hippocampal ensemble activity during rapid eye movement sleep. Neuron. 2001 Jan;29(1):145-56. PubMed.
- Stickgold R, Walker MP. Sleep and memory: the ongoing debate. Sleep. 2005 Oct;28(10):1225-7. PubMed.
Further Reading
News
- Does Amyloid Disturb the Slow Waves of Slumber—and Memory?
- Sleep Gene Switch Means Lights Out, Memory On, for Fruit Flies
- Remember, Remember…Growth Factor, Timing, and Sleep
- Sleep On It—Astrocytes May Play Key Roles in PD, AD
- Making and Mining Memories—New Insight Into Hippocampal Role
- Smell You Later—Odors Help Consolidate Memory Overnight
- Could Alzheimer’s Drugs Mean “Good Night” to Good Memory?