Off-target troubles aside, tau tracers took the cake at the 10th Human Amyloid Imaging conference, held January 13-15 in Miami Beach, Florida. Even as researchers continue to sort out the vagaries of working with investigational tracers, they have started delving into deeper questions than whether neurofibrillary tangles are merely present in the brain. Scientists are asking how patterns of tau deposition relate to Aβ, neurodegeneration, and connectivity in Alzheimer’s disease. Preliminary cross-sectional data hint at a considerable lag between Aβ and tau accumulation in familial AD. New ligands debuted at HAI this year, but whether they will better detect tau deposits than the current batch remains to be seen. Leaders in the field still don’t know how well tau PET will work in other forms of dementia, such as FTD, PSP, and CBD.
Tau Takes Center Stage at 10th Human Amyloid Imaging Conference
Since it started in 2007 as a one-day powwow of 150 early aficionados in Boston, the annual Human Amyloid Imaging meeting has become a fixture for anyone using imaging technology to study dementia-related pathology in the brain. At the 10th meeting, held January 13-15 in Miami Beach, Florida, 2½ days of talks and ample discussion drew nearly 350 attendees, some spilling into overflow space. This year, the meeting focused overwhelmingly on tau PET imaging and its many challenges. HAI scientists debated whether the current batch of tau ligands are suitable to answer questions such as where, when, and in what form tau aggregates, and how those aggregates affect the normal working of the brain. For Alzheimer’s disease, scientists are beginning to make inroads into this territory. In the case of non-AD tauopathies, the jury is still out, as the ligands bind poorly to the various forms of tau found in these dementias, and specificity remains a concern (see Part 3). HAI is co-organized each year by Keith Johnson, Massachusetts General Hospital, Boston; William Jagust, University of California, Berkeley; and William Klunk and Chet Mathis of the University of Pittsburgh.
Location Matters, But What’s With the Inferior Temporal Lobe?
In their effort to understand how Alzheimer’s develops, scientists are now using multimodal imaging to relate tau aggregation with structural and functional changes. Molly LaPoint, working with Johnson and Reisa Sperling at Brigham and Women’s Hospital, Boston, examined the relationship between accumulating tau in various regions of the brain and thinning of the cortex. LaPoint analyzed data from 99 normal older individuals in the Harvard Aging Brain Study (HABS). She identified 68 regions of interest (ROIs), 34 per hemisphere, for analysis. Then she correlated how much the cortex had shrunk in thickness, measured by two to three MRI scans taken over approximately three years, with how much of the tau tracer AV1451 those 68 areas had taken up in a PET scan done close to the volunteer’s last MRI. LaPoint looked for correlations between AV1451 binding and thinning within the same ROIs, on the one hand, and between tracer uptake specifically in the inferior temporal lobe with global cortical thinning elsewhere, on the other.
Tau and Thinning.
More cortical thinning (shown) occurs when neurofibrillary pathology is high in the inferior temporal lobe. [Image courtesy of Molly LaPoint.]
In a cross-sectional analysis of the whole cohort, LaPoint found that, for many brain regions, AV1451 uptake correlated with thinner cortices in the same region. The correlation was highest in the temporal lobe, where tangles are known to appear earliest as people age.
Looking at thinning over time, LaPoint found similar relationships among several ROIs mostly in the right hemisphere, including the right superior temporal gyrus, right middle temporal gyrus, and the right temporal pole; all were thinning faster with higher tau.
But it was not just local tau that correlated with a thinning cortex. Though less widespread, a similar pattern emerged between AV1451 uptake in the inferior temporal lobe and thinning in medial and lateral temporal lobe regions. Furthermore, higher AV1451 binding in the inferior temporal lobe also was linked with thinning of the right middle temporal, right fusiform, right anterior cingulate, and right and left parahippocampal gyri. Curiously, for many of these regions, thinning more strongly related to tau in the inferior temporal lobe than to local tau.
Researchers at HAI intensely discussed this asymmetrical pattern, particularly the apparent remote influence of inferior temporal lobe tau. Clifford Jack, Mayo Clinic Rochester, Minnesota, wondered why the correlations were tighter in the right hemisphere. After all, most people in this cohort would not be expected to be on their way to disease with such asymmetry. Sperling noted that everyone entering HABS must have a clinical dementia rating of zero, meaning that candidates with left cortical thinning who performed poorly in memory tests would have been excluded.
Toxic Tau.
Modelling predicts that higher tau tracer uptake means faster thinning of the parahippocampal gyrus. [Image courtesy of Molly LaPoint.]
Some wondered why tau in the inferior temporal lobe seemed to hold more sway over cortical thickness than did local tau. Jagust asked if tau in the inferior temporal lobe exerts remote effects, or if its presence in this region merely signals that atrophy is afoot elsewhere in the brain. “In our view, it is mostly the latter,” said Johnson. Jagust then wondered if tau in other regions of the brain affects disease. “Many of us felt that, unlike Aβ, tau would help us understand why some regions are vulnerable and others aren’t,” he said, asking, “Can you predict atrophy by knowing where tau is? Or is it not important where tau is?” LaPoint cautioned that these are normal elderly whose pathology was perhaps insufficient to draw out such relationships.
Mark Mintun of AVID Radiopharmaceuticals in Philadelphia noted that in symptomatic stages of the disease, the distribution of tau deposits does match the regional dysfunction predicted by cognitive testing, suggesting that local tau is important. Jagust agreed that this is true in later stages, but said he was more interested in how the disease develops and spreads. “In late disease Aβ is everywhere, but there are focal effects. How does the spread of tau fit in?” he asked. Johnson noted that LaPoint was looking at large ROIs. “We may need to look at tighter distribution and spread of tau to make those correlations,” he said.
Functional Connectivity and Temporal Lobe Tau
Other researchers are studying functional networks to understand the consequences of where tau deposits. Aaron Schultz from Massachusetts General correlated AV1451 uptake with activity in four canonical cortical networks—default mode (DMN), ventral attention (VAN), dorsal attention, and frontoparietal control. Among 103 participants in the HABS, a correlation emerged between inferior temporal AV1451 and connectivity, but only among those 29 subjects who also had a positive Aβ scan as per PiB PET. Specifically, inferior temporal tau came with decreased connectivity in the VAN and DMN networks; the other two networks trended toward reduced connectivity as well. Curiously, tau in the entorhinal cortex—a brain region widely studied for its tau burden in many human and mouse studies—appeared unrelated to connectivity. This again pointed to something unique about the inferior temporal lobe and its ability to influence other areas of the brain.
When Shultz compared connectivity in people with different amounts of amyloid and tau pathologies, he found an inverted-U-shaped response. Low amyloid and tau predicted low connectivity across the VAN and DMN networks; medium levels of amyloid and tau came with high connectivity, and peak levels of Aβ and tau pathology once again came with reduced connectivity. This up-down curve may help explain conflicting reports variously linking amyloid to increased or decreased functional connectivity in preclinical groups, said Schultz.
Researchers at HAI wondered if amyloid contributes to the connectivity changes more than tau. Schultz believes tau pathology plays a key role because, in its absence, the functional connectivity correlations disappear. Gil Rabinovici, University of California, San Francisco, noted that Schultz had measured AV1451 uptake in a node of the brain that lies outside the affected networks, and wondered if tau in other regions might correlate better. Shultz next plans to tease out local disruptions, and how tau spread might affect connectivity.
What Does Tau Have to do with Atrophy, Functional Connectivity?
Possible damage from local tau emerged in functional analyses by Rik Ossenkoppele, University of California, San Francisco. Ossenkoppele reasoned that if tau deposition spread along functional networks, its distribution pattern in a given person should reflect the areas of the brain most affected by that person’s specific clinical form of dementia. For example, patients with posterior cortical atrophy (PCA) tend to have visual impairments, suggesting visual network dysfunction, while people with logopenic variant primary progressive aphasia (lvPPA) struggle to find words and parse sentences, indicating problems in their language centers.
Telltale Troika. In AD dementia, tau covariance (top) based on right middle frontal gyrus atrophy (middle) overlaps with functional connectivity maps of the right executive control network (bottom). [Image courtesy of Rik Ossenkoppele.]
Working with Rabinovici at UCSF and others, Ossenkoppele measured AV1451 uptake in eight people with lvPPA, seven with PCA, five with late-onset AD, and seven with early onset AD. All had brain Aβ as per PiB PET, except one lvPPA patient whose amyloid status was unknown. Ossenkoppele generated maps of tau covariance based on a method originally devised by William Seeley and colleagues (see Apr 2009 news). The idea is that when correlations of tau uptake among different brain regions hold up between different people, they are highly “covariant,” and therefore likely meaningful. The image above, for example, shows that uptake of AV1451 in three voxels varies in the same way, i.e. it covaries, across seven people (S1 to S7). With this voxel-wise approach, Ossenkoppele correlated tau uptake in four seed regions with uptake throughout the brain and across the 21 dementia patients. The starting seeds were in the right middle occipital gyrus, the left superior temporal gyrus, the right middle frontal gyrus, and the left posterior cingulate. He chose the first three because prior work indicated they correspond to areas where the brain shrinks most in PCA, lvPPA, and AD, respectively (see Migliaccio et al., 2009). He chose the left posterior cingulate as a common denominator because it shrinks in people who have any of these diseases.
Ossenkoppele compared how patterns of tau covariance matched up against eight known cognitive networks that had been previously established in 1,000 young adults. They are the higher visual, language, salience, sensorimotor, right and left executive control, and ventral and posterior default mode networks. The overlap was striking and supports the idea that tau deposits spread along functional networks in the brain. For each of the four atrophy seeds, the tau covariance matched the connectivity networks affected by the given disease. PCA, for example, affects the visual network, and this network in turn maps closely to tau covariance with the right middle occipital gyrus, which atrophies most in that disease. Likewise, tau patterns matched networks affected in lvPPA and early onset AD. Tau covariance with the right middle frontal gyrus overlapped with the right executive control network that atrophies in typical AD, while tau covariance with the left superior temporal gyrus seed, which atrophies in lvPPA, overlapped with the language network. Lastly, the left posterior cingulate tau covariance matched the posterior DMN, a region susceptible to Aβ deposition in all forms of AD.
Others praised the approach, and indeed it earned Ossenkoppele the 2016 HAI Young Investigator Award. Sperling wondered if testing covariance based on peak areas of AV1451 rather than atrophy might reveal a different picture. Ossenkoppele said he chose atrophy seeds because he wanted to look at variance by disease phenotype, but also because he wanted to avoid bias. “We preferred to select the seeds from an independent sample of patients and we feel that’s a strength of the study,” though he agreed with Sperling that analyzing covariance based on peak tau uptake would be another way to parse the data, he told Alzforum.
Jack wondered if the data could be explained by local vulnerability to tau pathology. Rabinovici noted that tau covariance with the language and executive control seeds was not contiguous, but jumped between separate areas that are functionally connected. This hints that tau spreads along neuronal projections that make up functional networks. “We need longitudinal analysis to address that question,” he said.
Confused? It gets worse: David Jones, Mayo Clinic, Rochester, Minnesota, cautioned that expression of different genes covaries in these networks, as well, and this may further complicate the interpretation of how tau spreads.—Tom Fagan
Tau Tracers Track First Emergence of Tangles in Familial Alzheimer’s
PET imaging transformed Alzheimer’s research when it enabled scientists to track the spread of Aβ pathology in living people. It revealed that amyloid plaques appear decades before symptoms of Alzheimer’s disease emerge. But what about neurofibrillary tangles? With the advent of tau tracers, scientists are now beginning to correlate the two major hallmarks of AD in real time. The results may clarify exactly how AD develops before a patient notices that anything is amiss. At the 10th annual Human Amyloid Imaging meeting, held January 13-15 in Miami Beach, Florida, preliminary data from two autosomal-dominant AD (ADAD) cohorts suggest that neurofibrillary tangles begin to accumulate much closer to the age of onset of symptoms than do amyloid plaques. “The data seem to suggest that in familial disease, tau tangles emerge long after Aβ but then rapidly spread,” said Reisa Sperling, Brigham and Women’s Hospital, Boston, a lead investigator on one of the studies. In short, when the tangles come, the real trouble starts.
Familial vs. Sporadic. Tau pathology appears more widespread in autosomal-dominant (left) than sporadic AD. [Image courtesy of Tammie Benzinger.]
Scientists are curious to see how tangles develop in autosomal-dominant AD because patients are much younger at age of onset than those who develop sporadic AD, and likely have less age-related tau accumulation. Sperling collaborates on this study with Francisco Lopera and colleagues at the Universidad de Antioquia, Medellin, Colombia; Eric Reiman at Banner Health, Phoenix; and Yakeel Quiroz and Keith Johnson and colleagues at Massachusetts General Hospital. The scientists have scanned volunteers from a kindred in Colombia that lives with the presenilin1 E280A, aka Paisa, mutation.
Similarly, researchers led by Tammie Benzinger at Washington University, St. Louis, have been imaging tau deposits in families from the multinational DIAN observational study of familial AD. In her talk, Benzinger outlined early results for a group of 16 DIAN volunteers, both carriers and non-carriers of an FAD mutation. Because this sample size was so small, the DIAN study rules preclude divulging many details of these volunteers for fear of accidentally “unblinding" their genetic status and revealing private information. Benzinger presented just enough information to draw some tentative conclusions.
Benzinger compared the pattern of tau deposition in these DIAN volunteers with that seen in sporadic AD. Of the people so far scanned for tau, only two were classified as positive for tangles based on their uptake of the PET tracer AV1451. While the regional distribution of uptake in their brain was about the same as that seen among patients with sporadic AD, the amount seemed higher in the ADAD patients, Benzinger said. Both these DIAN participants were very mildly symptomatic, yet they had strikingly more AV1451 retention in the precuneus, parietal, and frontal lobes than sporadic patients with the same level of impairment, said Benzinger.
This might suggest more aggressive tau deposition in familial than in sporadic AD. However, that tau might be coming on quite late. The two tau-positive volunteers were within a few years of their expected age of onset. One cognitively normal volunteer with widespread Aβ deposition based on a PiB PET scan had a completely normal AV1451 scan. This person was 10 years younger than their predicted age of onset. By comparison, Aβ begins to deposit in the brain as much as 30 years before symptom onset.
For its part, the Colombian study suggests a similar timeline for tau. Analysis of the few scans done thus far hints that people with the Paisa mutation must be within about six years of age of onset before tau accumulates to detectable levels. In her poster, Quiroz described how she correlated PiB and AV1451 uptake in six asymptomatic PS1 mutation carriers and six age-matched non-carriers who last summer travelled from Colombia to Boston to be scanned. While the mutation carriers had higher cortical PiB uptake than same-age controls, no significant difference between the groups was evident on their AV1451 scans. Uptake of the tau ligand in the inferior temporal lobe and entorhinal cortex trended higher in mutation carriers than controls, as did an association between AV1451 uptake and amyloid burden. However, the latter trend was driven solely by one older mutation carrier, who was six years away from predicted age of onset in this cohort. The other five carriers were around age 30.
Sperling suggested that these patients’ relative youth may make them more resilient and that is why tangles do not emerge until fairly close to disease onset. But since FAD patients have more tau by the time mild symptoms start than do sporadic patients, their tau deposition must really ramp up quickly, she said. Quiroz agreed, and noted that age may play a role. “When thinking about sporadic AD, we have to consider that aging may confound the data,” she said. “People in their 70s already have tau pathology and have had it for many years, so some of the accumulation prior to onset is age-related.” That doesn’t happen with ADAD because the people are very young.
Both familial studies also suggest a lag between tau turning up in the cerebrospinal fluid and tangles forming in the brain. Previously WashU’s Ann Fagan and the DIAN team reported that CSF tau rises about 15 years before age of onset (Bateman et al., 2012). Last year Quiroz and colleagues reported the same in the Colombian kindred (see Jan 2015 news).
“We see the same mismatch in sporadic AD,” Benzinger told Alzforum. “I think we may ultimately decide that CSF and PET imaging are measuring two different aspects of tau pathophysiology.” She suggested that CSF tau may represent neuronal injury, which is why there are transient fluxes/increases in CSF tau in other instances of injury, such as trauma or stroke, while imaging captures the location of the abnormally folded neurofibrillary tangles. Quiroz has CSF samples from the 12 patients she scanned but has yet to run the analysis. She has applied for funding to run tests every 18 months on 30 mutation carriers and 30 controls.—Tom Fagan
Bateman RJ, Xiong C, Benzinger TL, Fagan AM, Goate A, Fox NC, Marcus DS, Cairns NJ, Xie X, Blazey TM, Holtzman DM, Santacruz A, Buckles V, Oliver A, Moulder K, Aisen PS, Ghetti B, Klunk WE, McDade E, Martins RN, Masters CL, Mayeux R, Ringman JM, Rossor MN, Schofield PR, Sperling RA, Salloway S, Morris JC.
Clinical and biomarker changes in dominantly inherited Alzheimer's disease.
N Engl J Med. 2012 Aug 30;367(9):795-804.
PubMed.
Further Reading
No Available Further Reading
Shaky Specificity of Tau PET Ligands Stokes Debate at HAI
The emergence of PET ligands for tau has the potential to revolutionize the study of the whole gamut of tauopathies. Researchers are tracking how neurofibrillary tangles appear and spread in real time, yielding invaluable insight into the pathological course of Alzheimer’s disease. For other tauopathies, however, the scans seem open to interpretation. As discussed at the Human Amyloid Imaging meeting, held January 13-15 in Miami Beach, Florida, the problem boils down to two properties biologists constantly bump into: sensitivity, i.e., how little can be detected, and specificity, i.e., lack of off-target binding. At this early point in this rapidly evolving field, it appears that the tau ligands available thus far poorly bind to the particular types of fibril that form in frontotemporal dementia, progressive supranuclear palsy, Pick’s disease, and other non-AD tauopathies. Moreover, the ligands appear to bind to other things as well. Complicating the issue even further, the ligands perform a “now you see me, now you don’t” act that has scientists scratching their heads.
Does It, or Doesn’t It? Maybe It’s the Assays …
The tau ligands’ penchant for prestidigitation was unveiled in Marta Marquié’s talk. Last year, Marquié, who works with Teresa Gomez-Isla at Massachusetts General Hospital, Boston, reported that the PET ligand AV1451 bound to tissue sections from AD brains, but not to tissue taken from people who had had other tauopathies, such as progressive supranuclear palsy (PSP), corticobasal degeneration (CBD), or Pick’s disease (Marquié et al., 2015). AV1451 did, however, appear to visualize tau in vivo in those tauopathies. How could that be? This year at HAI, Marquié addressed this question by correlating in vitro binding directly with in vivo imaging data. She reported a detailed postmortem analysis of tissue samples taken at autopsy from two patients who had non-AD dementia and had had a tau PET scan within eight months of death. “These are the first autopsy-confirmed non-AD tauopathy cases imaged while alive that have been reported,” Gomez-Isla said.
One was a 56-year-old man who carried the P301L tau mutation and had been diagnosed with behavioral variant FTD. He had atypical parkinsonism and had been declining for 2.5 years. PET imaging with AV1451 showed the ligand bound predominantly in his basal ganglia and midbrain. Gomez-Isla noted that in vivo uptake of this tracer in midbrain and basal ganglia has been noticed in a high percentage of cognitively normal older adults who are not expected to harbor tau lesions in those regions. This man had much less AV1451 uptake in the inferior temporal and frontal cortex, where tau deposits in bvFTD. In contrast, his autopsy revealed tau-positive grains not only in the basal ganglia and white matter, but also extending into the cortex.
What was the ligand binding to? Marquié used phosphor screen and nuclear emulsion autoradiography to compare AV1451 binding to tissue slices with tau staining by immunohistochemistry. In short, the in vivo and in vitro signals only partially correlated. Marquié found no AV1451 binding in slices from any brain region except the entorhinal cortex and the substantia nigra. The latter matched with the live scan, but the former did not. Age- rather than disease-related tau deposits probably explain the entorhinal cortex binding, said Marquié. She noted this binding could serve as an internal positive control. Neuromelanin, which AV1451 is purported to bind, may account for some of the signal in the substantia nigra. Marquié interpreted the results to mean that while AV1451 binds to the paired helical fragments of tau found in Alzheimer’s disease, it does not bind the straight tau filaments known to occur in bvFTD and other non-AD tauopathies with an affinity that could explain in vivo signals.
Analysis of tissue from the second patient supported this idea. This 68-year-old man had a neuropathologically confirmed diagnosis of progressive supranuclear palsy (PSP). Similar to the bvFTD case, Marquié detected no lifetime AV1451 PET binding to any of the regions suspected of having high tau pathology other than the basal ganglia and midbrain. Immunohistochemical analysis of sections of his brain tissue, using antibodies that recognize tau pathology in PSP, detected abundant tau pathology in the brain stem, cerebellum, basal ganglia, midbrain, and numerous other regions. Once again, autoradiographs of brain slices showed AV1451 binding to only the entorhinal cortex and substantia nigra.
“Our autoradiography studies have demonstrated that the nearly universal midbrain in vivo signal seen in older adults, regardless of the presence or absence of tau pathology, is very likely due to off-target binding of this tracer to neuromelanin-containing neurons in the substantia nigra,” Gomez-Isla wrote to Alzforum. She does not know what gives rise to uptake of AV1451 in the basal ganglia in vivo, and did not detect any binding in this area by autoradiography or by in vitro binding assays. “That’s what makes us think that it could be, at least in part, a non-specific in vivo uptake related to technical or biological factors other than tau pathology itself,” she wrote.
Milos Ikonomovic, working with HAI co-organizer William Klunk at the University of Pittsburgh and collaborators, drew similar conclusions about AV1451 from his ex vivo binding studies. Ikonomovic compared ligand binding in brain tissue from three people who had had PSP, two who had had CBD, and one with Pick’s disease; he used two AD cases with confirmed tau pathology as positive controls. He also had brain slices from three cases of FTLD with TDP-43 inclusions—a tau-negative form of dementia. Both tritiated and F18-labelled AV1451 bound strongly to frontal cortex in AD samples but weakly or below control levels in frontal cortex from the other tauopathies, even though immunohistochemistry and ELISAs detected much more tau pathology in Pick’s disease and CBD than in PSP and FTLD-TDP. “We believe that the tau aggregates that form in non-AD tauopathies are distinct from the paired helical fibrils that form in AD,” said Ikonomovic. He added that this could explain why tau ligands poorly bind in non-AD tauopathies
These presentations prompted a searching discussion of whether the in vivo PET imaging or the autoradiography and other in vitro binding assays were more likely to be correct. Interestingly, Marquié found that in vivo AV1451 uptake in the pallidum, a part of the basal ganglia, correlated with tau levels there as measured by semi-denaturing detergent acrylamide gel electrophoresis. SDD-PAGE is gentle enough to leave amyloid-like polymers intact. “The correlation of AV1451 signal with tau levels measured by SDD-PAGE in pallidum in this case matches our in vivo findings,” noted Gil Rabinovici, University of California, San Francisco. “This highlights the globus pallidus as the area of highest signal, and supports the notion that this signal may be related to tau,” he said.
Attendees spent considerable time debating why the autoradiographs are turning up negative. “It is important to recognize that these assays are sensitive to experimental conditions,” said Rabinovici. In another presentation, Giorgio Attardo from Avid Radiopharmaceuticals, the supplier of AV1451, noted that autoradiograph protocols usually include a 70 percent ethanol wash, which may be too harsh. Attardo used signal matching techniques to correlate AV1451 binding with AT-8 tau antibody immunoreactivity. This technique hinges on using the identical tissue slice for both autoradiography and immunohistochemistry. Attardo found that the two signals overlapped well in brain tissue from AD patients, with area correlations ranging from 88 to 92 percent, while this correlation was at best 26 percent in tissue from PSP patients and 63 percent in tissue from Pick’s disease patients. Leaving out the ethanol wash led to muddled autoradiogram tests of normal tissue sections, Attardo said, but reducing the ethanol to 30 percent revealed a second binding site that could be blocked by non-radiolabeled AV1451. “It may be that a relatively low affinity binding of AV1451 to tau aggregates in PSP is washed out during tissue preparation,” said Rabinovici.
Klunk noted that the harsh washing conditions were needed because AV1451 is such a sticky compound. “It could be, then, that all we can expect to detect in vitro are the tangles in AD,” he said. That is because AV1451’s affinity for those tangles is much higher. Klunk added that the field remains challenged to figure out if tracers are being artifactually removed during in vitro biding assays, or if the signal in vivo is spurious. “I think our current assays and current compounds don’t allow us to fully answer these questions,” he told Alzforum.
Others suggested measuring the specificity of these ligands by running a competition assay in vivo with unlabeled ligand. That led to nods of support, but some quick mental math by HAI co-organizer Chet Mathis, University of Pittsburgh, squashed the idea, for AV1451 at least. “The dose is the problem,” Mathis said. “We would need several hundred milligrams of cold AV1451, and I doubt the toxicity and solubility would allow it.” Mathis suggested testing this in animal models. Perhaps such a “cold” competition assay might help identify non-tau binding sites? In his presentation, David Jones, Mayo Clinic, Rochester, Minnesota, reported that people with a form of FTD caused by progranulin mutations bind AV1451 in the brain. As Reisa Sperling of MGH noted, those patients do not have tau deposits on autopsy (see Part 4 of this series). One idea is that AV1451 might bind monoamine oxidase and, in fact, Attardo found that the MAO inhibitor clorgiline blocked AV1451 binding in slices of striatum. Clorgiline or isocarboxazid, another MAO inhibitor, might be safely given in sufficient doses to block AV1451, Attardo said.
Finally, some researchers raised the tantalizing possibility that AV1451 and other tau ligands might be binding something altogether different. Even if that something is not tau, it could be informative. Almost no off-target binding of AV1451 occurs in 30-year-olds, for example, implying that the target is not something normally abundant in the brain. “That, plus Jones’ data on FTD, suggests to me that AV1451 might bind some other marker of neurodegeneration,” said Sperling.—Tom Fagan
At HAI, Researchers Explore Diagnostic Potential of a Tau Tracer
The accumulation of tau protein in the brain reflects a spectrum of different human neurodegenerative diseases, from those affecting the front of the brain, such as frontotemporal dementia, to those affecting the back, such as posterior cortical atrophy. Scientists are hoping that tau PET imaging will pinpoint those deposits in living people with sufficient accuracy to radically improve the diagnosis, prognosis, and basic understanding of these tauopathies. Alas, this vision may take a bit longer to realize. At this year’s Human Amyloid Imaging conference January 13-15 in Miami Beach, Florida, scientists reported how the leading tau ligand in many cases falls short of distinguishing people with disease from normal healthy controls. HAI made abstracts and keynote talks from Henrik Zetterberg, University of Gothenburg, Sweden, and Charles Duyckaerts, Hôpital Pitié-Salpêtrière, Paris, available here.
Separating Disease from Controls
PET imaging has revolutionized the study of amyloid plaques, and scientists are hoping it will do the same for tau. In the case of Alzheimer’s at least, it may. Neurofibrillary tangles that emerge as AD progresses seem to avidly bind several tau ligands currently in development, and researchers are using tau PET to get a better handle on how Aβ and tau conspire to create havoc in the brain (see Part 1 of this series). As noted time and again at HAI, however, non-AD tauopathies are a different kettle of fish. Researchers noted that while AV1451 seems to bind where one might expect in a given case of tauopathy, it falls short when it comes to being diagnostically useful.
Telltale Tangles.
In one tau mutation carrier, AV1451 binding in the frontal cortex suggests pure frontotemporal dementia (top), while in another PET signals in posterior areas correlate with concomitant Aβ amyloidosis (bottom). [Courtesy of Richard Tsai.]
Richard Tsai, who works with Gil Rabinovici and others at the University of California, San Francisco, examined AV1451 scans from volunteers with different tauopathies and clinical symptoms. His sample included four people with tau mutations, three with a clinical diagnosis of the nonfluent variant of primary progressive aphasia (nfvPPA) and five with corticobasal syndrome (CBS). While the distribution and strength of the tau signal varied, by and large the ligand bound to regions affected by ongoing disease in each case, said Tsai. Interestingly, the imaging brought out subtle differences among the volunteers that fit with their individual pathologies.
For example, two people with different tau mutations bound the ligand in their frontal cortex, consistent with their frontotemporal dementia diagnosis. However, while clinicians gave both of these patients a clinical dementia rating of 2.0, the person carrying a V337M mutation had much higher uptake value ratios (SUVR) for AV1451. This could mean that this mutation leads to the type of paired helical filaments found in AD, said Rabinovici.
Did the PET scan of the other patient underestimate their tau burden? It turned out that this second person carried a P301L mutation, which typically forms straight filaments rather than the paired helical type. This person also had ongoing Aβ amyloidosis as judged by PiB PET. AV1451 bound farther back in the brain, in the parietal and occipital regions, indicating more widespread tau pathology such as might be found in typical Alzheimer’s and suggesting this was a case of mixed dementia.
Two asymptomatic volunteers carrying the same mutation in intron 10 of the tau gene had lower AV1451 uptake; this was consistent with the presence of straight filaments and respective CDR scores of 0.5 and 0.0.
Likewise, among the three nfvPPA patients, PET imaging correlated with subtle differences. Each had a negative PiB PET scan, and their MMSE scores of 26-28 suggested mild cognitive impairment. While all took up a similar amount of AV1451, its distribution varied. In two right-handed volunteers, tracer bound predominantly on the left side of the brain, while the one lefty had a more even distribution between the two hemispheres.
In CBS, AV1451 distribution reflected both right-left asymmetry and underlying Aβ pathology. Of the five patients Tsai scanned, one had a positive PiB PET scan. That person had rampant tau pathology throughout the cortex. Among the other four, AV1451 distributed through the dentate, putamen, pallidum, and white matter. More bound to the left hemisphere in three patients with predominantly right motor symptoms, and to the right hemisphere in a person with left-predominant motor symptoms.
Tsai’s work is good news to the field because it suggests that AV1451 binding reflects the distribution of tau pathology in individual cases of tauopathy. Does that make tau PET a diagnostic tool? It is too early to tell, scientists agreed. In all pure tauopathy cases scanned thus far, much less AV1451 bound in the brain than is typically seen in AD. The consensus at HAI was that the signal may be too weak to distinguish people with those tauopathies from people with age-related tau deposition.
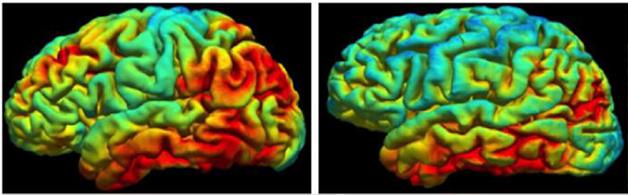
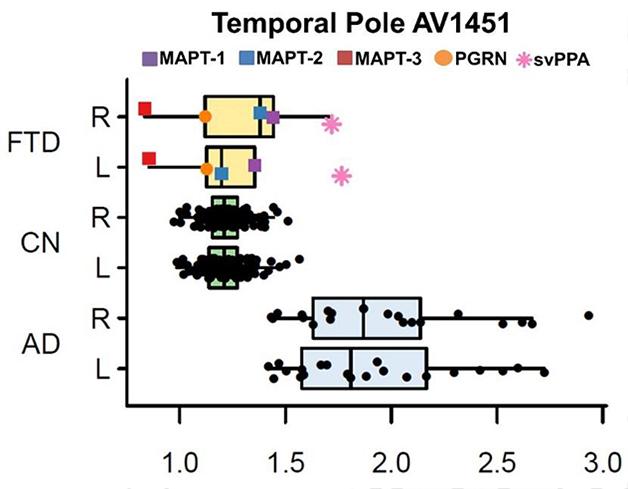
In his presentation, David Jones, working with Bradley Boeve and colleagues at the Mayo Clinic in Rochester, Minnesota, brought home the cold truth by comparing ligand uptake among a larger sample of cognitively normal controls and patients with AD and FTLD. Jones reported that both visual reads and SUVR quantitation of scans did indicate uptake of AV1451 in the brains of five people—three with tau mutations, one with a progranulin mutation, and one with semantic variant PPA. However, comparing their scans with those from 21 AD patients and 120 normal controls revealed that only the semantic variant case bound more ligand than the controls. For the other four patients, SUVRs in both temporal and frontal cortex overlapped with control values (see figure below). Jones therefore concluded that AV1451 binding is insufficient to distinguish FTLD cases.
In Alzheimer’s (AD), uptake of the tau tracer AV1451 is higher than in controls. In frontotemporal dementias (FTD) caused by different mutations, it overlaps with controls (CN). [Courtesy of David Jones.]
Off the Mark
Furthermore, Jones’ work emphasized how off-target binding might be a problem. Although FTD patients with progranulin mutations typically have deposits of TDP-43 rather than tau, the PGRN patient Jones scanned bound AV1451 in the frontal lobe. Since previous work suggests that AV1451 has no affinity for TDP-43 deposits (see Feb 2015 conference news), researchers were puzzled by this binding. Rabinovici, Cliff Jack from the Mayo Clinic, Rochester, Minnesota, and Reisa Sperling from Brigham and Women’s Hospital, Boston, all noted that it occurred in an area of ongoing neurodegeneration. They wondered if the ligand might be binding a marker of degeneration other than tau. Jones considered this possible, but would not speculate on what that may be. “We don’t yet understand if this is a non-specific measure of neurodegeneration,” Rabinovici told Alzforum. “There is more work to be done to understand the nature of that signal.”
Diagnosing progressive supranuclear palsy (PSP) based on tau PET may be an even tougher nut to crack. A number of companies are beginning to evaluate tau-based drugs in PSP and would welcome a clear-cut molecular imaging marker for this disease (see Therapeutics database). The problem is that in normal controls, AV1451 binds in some of the very same areas where tau deposits in PSP, including the midbrain and basal ganglia.
Scientists have not given up hope that they may yet tease out a disease-specific signal by homing in on certain regions of the brain. With this in mind, Daniel Schonhaut, who works with Rabinovici at UCSF, compared AV1451 scans of 17 PSP patients at three centers with those from 28 normal controls who tested negative on PiB PET, indicating they had no concomitant amyloid pathology that might complicate the results. Voxel-wise analysis brought out differences between patients and controls. In PSP, AV1451 binding in the pallidum, putamen, dentate nucleus of the cerebellum, and subthalamic nucleus was higher than binding in those regions in controls. In addition, binding in patients trended higher in the substantia nigra, thalamus, caudate, and pons.
The biggest difference occurred in the pallidum, where the SUVR averaged about 2.0, compared to 1.6 in controls. Schonhaut calculated that a threshold for the pallidum of 1.75 could give a sensitivity and specificity of 0.81 and 0.93, respectively. However, there was no correlation between AV1451 uptake and the PSP Rating Scale, which measures disease severity. Longitudinal data are needed to better tease out that relationship, said Schonhaut.
Schonhaut further claimed that AV1451 might prove useful in distinguishing different forms of PSP. Classic PSP, aka Richardson’s syndrome, affects a wider area of the brain, whereas a form called pure akinesia with gait freezing spares the dentate nucleus of the cerebellum. Diagnosing the latter is challenging because symptoms overlap with those of Parkinson’s disease and other dementias, said Rabinovici. Schonhaut reported that among three patients of each type, AV1451 uptake in the dentate nucleus matched their subtype. “While this is a small number of patients, the results are encouraging,” he said.—Tom Fagan





Comments
No Available Comments
Make a Comment
To make a comment you must login or register.