CONFERENCE COVERAGE SERIES
Neurobiology of Aging 2004
San Diego, California
21 – 22 October 2004
CONFERENCE COVERAGE SERIES
San Diego, California
21 – 22 October 2004
To the student of neurodegenerative diseases, the Society for Neuroscience conference can seem like an overloaded buffet where only morsels of the food tickle one’s taste buds. By comparison, the satellite meetings offer a carefully selected menu with ample time to consume and digest new findings in good company. This was true of the 5th Neurobiology of Aging Conference, held on October 21 and 22. Organizers Ralph Nixon and Paul Coleman chose protein misfolding in Alzheimer’s and other age-related neurodegenerative diseases as the topic, and if full attendance and lively debate at the end of two intense days is any indication, this small conference was a resounding success.
Nixon, of New York University’s Nathan Kline Institute in Orangeburg, opened the meeting with a challenge. A growing number of scientists believe that the accumulation of incorrectly folded proteins is a molecular basis of many diseases, including Alzheimer’s, Huntington’s, Parkinson’s, prion diseases, as well as peripheral conditions and even some forms of cancer. The study of both protein misfolding and of defects in the cell’s protein disposal systems has come a long way in the last 15 years. Even so, knowledge gaps remain wide and deep, Nixon said. For one, whatever the investigators' favored pathogenic hypothesis, their talks tend to leave obscure how a given misfolded protein kills the cell. “Something happens to the proteolytic system under study, then comes a black box, then comes cell death. We need to know who the killers are and how the cells are dying,” said Nixon. For another, it remains unclear whether misfolding effects precede or follow synaptic deficits in AD pathogenesis. Consensus disease pathways remain an elusive wish for all of the major neurodegenerative diseases. To try to bridge those gaps, Nixon rallied the audience to speculate beyond their data and shake up collective thinking.
Approaching the problem from many different angles, the talks brought to mind the adage of the blindfolded scientits, each of whom touches a different part of the elephant and proclaims it to be a tree, a snake, or something else. The discussions did not quite gel into a clear picture of the elusive elephant, either, but as the next best thing, here are the component themes that resonated throughout the event and drew some consensus at the end:
What therapeutic prospects emerge from recent advances on protein misfolding in neurodegenerative disease? Here, again, are common themes and caveats:
Below are summaries of selected talks at this conference, and more discussion of the therapeutic ideas.
Speakers include:
Up first was Fred Van Leeuwen from the Netherlands Institute for Brain Research in Amsterdam, who updated his hypothesis about molecular misreading and the ubiquitin proteasome system in neurodegeneration. The hypothesis proposes that frameshift transcription errors that occur continuously at a low level generate altered forms of certain proteins, including APP and ubiquitin B, which then contribute to the pathogenic process (see ARF related news story and Van Leeuwen et al., 1998). In the case of ubiquitin B, the altered form, UBB+1, cannot perform its normal function of ubiquitinating target proteins for degradation in the proteasome; instead, it becomes ubiquitinated itself. The resulting UBB+1-ubiquitin complex resists deubiquitination and could inhibit the proteasome (Lam et al., 2000). UBB+1 proteins occur in a variety of neurodegenerative diseases involving Aβ, tau, and huntingtin, but not α-synuclein (see De Pril et al., 2004 and Fischer et al., 2003).
At high doses, this protein inhibits the proteasome and causes cell death in culture. Its relevance in vivo, especially at low doses, has remained uncertain. Van Leeuwen addressed this issue by presenting initial data on transgenic mice that express UBB+1 transgenes postnatally in cortex and hippocampus. Both brain regions readily degrade low concentrations of UBB+1 (see Fischer et al., Soc. for Neuroscience, 2004 Abstract 336.1). They do not degrade high concentrations, however. By nine months of age, the high-expressing line of transgenics perform poorly in the Morris water maze and show proteomic changes downstream of UBB+1, (Van Dijk et al., Soc. for Neuroscience, 2004 Abstract 336.2.) This strain effectively represents a model for chronic proteasome inhibition, van Leeuwen added. Interestingly, a rat model of proteasome inhibition has generated recent interest in the Parkinson’s field because it mimics the disease more faithfully than do older models (see ARF related news story). Yet the comparison goes only so far, as the rats develop Lewy bodies, but Van Leeuwen’s group does not see UBB+1 and accompanying proteasome dysfunction in α-synuclein diseases.
Discussion after the talk centered on the question of whether UBB+1 accumulation and ensuing proteasome dysfunction is the first pathogenic event in AD, or it marks an earlier, unknown insult. Van Leeuwen noted that high concentrations of UBB+1 are required for proteasome inhibition, suggesting that other upstream compounds such as the ubiquitin-conjugating enzyme E2-25K/Hip-2 may be involved (Song et al., 2003). Clearly, Alzheimer disease is multifactorial, Van Leeuwen noted. Nixon asked what happens in the cell between proteasome inhibition and death? Attendants shared a sense that the proteasome field has not yet seen enough convergence and confirmation of data to allow conclusions about whether it plays a primary or secondary role in the disease process (for a review on this question, see Ciechanover and Brundin, 2003).
Ted Dawson illustrated one example of how emerging data are contradicting earlier, simpler hypotheses about the role of the proteasome in neurodegeneration. In Parkinson’s research, the spotlight fell on proteasomal degradation when it turned out that the parkin protein has an E3 ligase function (Shimura et al., 2001 and Zhang et al., 2000). Soon after that, potential substrates and interactors for parkin became known, and now the list includes synphilin-1, α-synuclein, CDCrel-1, p38, Pael-R, cyclin E, α/β-tubulin, synaptotagmin 11, among others. Researchers in Dawson’s lab at Johns Hopkins University in Baltimore are systematically testing which of these are relevant substrates in vivo, and whether their parkin-mediated degradation in the proteasome has an effect on disease. As a first step in this effort, Dawson reported that his group bred their parkin knockout mice (Von Coelln et al., 2004) to a strain transgenic for mutant human α-synuclein (Lee et al., 2002). Dawson reasoned that if parkin detoxifies α-synuclein via the proteasome, then its absence should worsen the phenotype of the α-synuclein mice. Alas, it did not. “We can take α-synuclein off the list of important parkin substrates,” Dawson said.
Next, Dawson tested synphilin-1, a parkin substrate and α-synuclein interactor that his group discovered (Chung et al., 2001). He noted that parkin does interact with synphilin-1, but that, at physiological concentrations, parkin appears to form alternate ubiquitin chains on amino acids other than the most studied one, lysine 48. In addition to tagging onto synphilin-1 conventional ubiquitin chains that lead to degradation, parkin also forms chains on lysine 63. These chains serve an unknown physiological function and lead to Lewy body formation, Dawson said. Taken together, Dawson does not view parkin as acting strictly in proteasome degradation. Instead, he believes it is a broad-spectrum neuroprotective agent, whose functions are determined in part by poorly understood modifications such as s-nitrosylation. —Gabrielle Strobel.
To be continued Friday, 17 December 2004.
No Available Further Reading
Speakers include:
Harald Janovjak
Eckhard Mandelkow
Judith Frydman
James Bowie
Charlie Glabe
What new techniques have come on line to study protein misfolding? Harald Janovjak introduced the audience to the latest refinements of atomic force microscopy, which make this desktop structural biology instrument ripe for attempts to image membrane proteins in neurodegenerative disease. Janovjak, a Ph.D. student at the University of Technology in Dresden, Germany, recapped his talk from a recent conference in Switzerland, and added further detail and perspective. For example, to illustrate how it is possible to correlate polypeptide sequence elements to unfolding, he ran a movie that showed the consecutive unfolding of pairs of helices in a single bacteriorhodopsin molecule pulled by the AFM tip.
Janovjak also talked about the AFM’s ability to detect assembly of protein subunits into functional complexes, a topic of interest in investigations of γ-secretase and APP. The AFM can contribute a variety of measurements to the understanding of protein misfolding, Janovjak said. For example, it can quantify rates of spontaneous misfolding, or probe parameters such as energy landscape, stability, and interaction with environmental components in different conformations of a given protein.
Eckhard Mandelkow, at Hamburg’s Max-Planck Institute in Germany, posed the problem of how one defines misfolding in a protein that is natively unfolded? To date, more than 100 proteins are known to fall into this category, including tau, Aβ and α-synuclein. A natively unfolded protein does not assume an ordered structure. Instead, it is flexible and becomes ordered only upon binding, i.e., when it performs a function, or indeed when it aggregates.
In the Aβ field, the argument regularly comes up that if only the physiological function of Aβ were known, its role in AD would become clear. Ironically, tau demonstrates that this is not necessarily the case, because its function is understood but its role in AD is not. Mandelkow said that tau’s role in axonal transport holds the clues to what it does in AD. Too much tau covers up the microtubules, blocking the movement of vesicles and organelles along microtubules, while too little tau destabilizes the microtubules (see ARF related news story). The balance of tau at microtubules is influenced by the concentration of soluble tau in the neuron, which, in turn, is one of the factors that determine its aggregation. Even so, how aggregation follows from excess soluble tau, and how this relates to axonal transport changes in AD remain a mystery.
One piece of the puzzle may lie in MARK kinases, which regulate tau levels on microtubules (see ARF related news story and ARF news story). Several different kinases can phosphorylate tau at different sites, but to date no consensus exists on whether increased phosphorylation promotes the formation of tau filaments or not. Mandelkow then reviewed research establishing a working model for how tau aggregates, and his lab’s recent establishment of a screen for aggregation inhibitors. Ongoing work will test these inhibitors in a new, inducible transgenic mouse model of tau.
Judith Frydman, of Stanford University, talked about the basic mechanisms of protein folding in live cells. The chemist and Nobel Laureate Christian Anfinson believed that a protein finds its normal folding state by itself. This may be true for some proteins, Frydman said, but in general, folding is more complicated, especially in cells. Two abundant chaperone systems aid the proper folding of newly translated proteins. Hsp70 is a monomeric protein that exists in many homologs in different cellular organelles, and chaperonins are oligomeric, ring-shaped complexes of which the major one in eukaryotes is called TRiC/CCT.
To find out how newly translated proteins fold, Frydman’s group performed dilution experiments similar to what Anfinson had done in his time. In the test tube, their model protein (the firefly luciferase, which cannot fold by itself) took a half-time of eight minutes to fold when placed in a mammalian cell extract containing chaperones. However, in live cells, where the chaperonins wrap around the nascent luciferase protein right as it peels off the ribosome, luciferase folds four times as quickly and with different intermediate states. This suggests that the cell contains a structured chaperone network at the ribosomes, Frydman said.
In further studies of protein folding during translation, this time with the tumor suppressor von Hippel Lindau (VHL) protein, the VHL substrate required the cooperation of Hsp70 and chaperonin to fold properly. Its cancer-causing mutations allow Hsp70 binding but interfere with subsequent chaperonin binding. Indeed, a proteomic analysis of chaperone-mediated folding suggests that a large fraction of cellular proteins transit through chaperone systems in the course of their folding, using first one, then another.
Besides supporting the folding of new proteins, chaperones also rescue proteins denatured by stress. Is the latter just a variation of the former, whereby either a new protein or a stressed one can be the substrate for chaperones but the process is the same? This is still an open question, but Frydman’s current data suggest that the process is a different one. The eukaryotic cytosol, she believes, contains separate chaperone networks that handle protein synthesis and quality control.
Finally, Frydman addressed the question of exactly what chaperones might do with misfolded proteins. They could be passive, merely holding on to the protein until it refolds properly. Or they could be more active, even interacting with ubiquitin ligase to feed a protein that is beyond repair into the proteasome. “We know very little about this,” Frydman said. To approach this issue, her coworkers took natural VHL mutations that generate misfolded VHL protein. They expressed these mutants in different yeast strains, one that degrades the mutant and one that can’t. They found that the chaperonin TRiC, which folds de novo VHL inside its cylindrical chamber, does not participate in its degradation. VHL degradation did require Hsp90, a chaperone not involved in its de novo folding. Other chaperones assist in both folding and degradation. This suggests that folding of new proteins and quality control of damaged proteins fall to distinct, specialized sets of chaperones, Frydman concluded. For more, see Spiess et al., 2004.
Two back-to-back presentations, one on computational structure prediction and one on cell biology/electrophysiology, highlighted a curious new discrepancy about which structure the Aβ peptide might assume. As a structural biologist, James Bowie of the University of California, Los Angeles, has not previously worked on neurodegeneration but ran smack into AD research this year. It happened when Bowie’s group tested a deceptively simple algorithm they had developed to model how certain membrane proteins fold. This research grew out of a frustrated acknowledgement that, despite 40 years of trying, scientists still have not cracked the old problem of de novo structure prediction.
Far fewer membrane proteins than cytosolic proteins have yielded insight into their atomic structure, largely because membrane protein biochemistry is so complex. Bench science aside, however, mere computational prediction of how a protein will fold ought to be easier for membrane proteins than for proteins in watery solutions, Bowie said. That’s because in water, a protein can assume a huge number of conformations, and sorting through them to find the right one remains daunting. With a membrane protein, its sequence implies where its transmembrane regions are, and once scientists have packed these, the possible structures for arranging the rest of the polypeptide chain become greatly restricted, Bowie said.
In this spirit, his group developed a three-step algorithm for folding homo-oligomeric transmembrane helices. Starting with helices in random orientation, the program runs 200 iterations of a procedure that finds conformations of minimal energy determined by the protein’s many van der Waals forces. Next, the algorithm filters out asymmetric conformations, then it clusters similar structures together. The largest cluster contains the structure most likely to be the correct one. “If someone had told me this approach a while ago I would have dismissed it as too simple. But as it turns out, this works really well,” Bowie said.
Bowie presented examples of validated or predicted structures, for example for glycophorin, for an influenza virus proton channel, and for the H. pylorum cytotoxin VacA. Like many of the proteins in this study, VacA appears to form a pore, and the commonality between these proteins turned out to be a packing motif Bowie called a glycine zipper. “We think this is an important mode of creating channel structures,” he said. One in four membrane proteins contains at least one glycine zipper motif—too many to examine. A more stringent database search, for proteins that contain a single transmembrane helix and at least four glycine zippers in a row, dredged up Aβ, Bowie said, as well as major prion protein precursor, which is also thought to form channels.
The idea that misfolded pathogenic proteins form pores in neuronal membranes has been around in the Alzheimer’s, Parkinson’s, Huntington’s, and prion fields for a decade without garnering wide support (see Kawahara et al., 1997, Lin et al., 1997, Kagan et al., 2002, Lashuel et al, 2003, Lashuel et al., 2002, Hirakura et al., 2000, and Kourie and Henry, 2002). Bowie said that his lab first reproduced some of the earliest data reported on the topic (Arispe and Rojas, 1993). Next, scientists began testing their own hypothesis that the glycine zipper motifs drive formation of the purported channels. They mutated different glycine positions on Aβ, and initial results suggest that wild-type Aβ forms channels, but the mutants do not, Bowie said. The mutants also appear less toxic to cultured neurons, but that work is even more preliminary. Bowie cautioned that he has no evidence as yet on whether his work is relevant to Alzheimer disease, and invited the field to come up with ways of finding out.
Ironically, Bowie’s research opens up a fresh vein of support for the channel hypothesis just as prominent work in the AD field appears to weaken it. This website has reported extensively on Charlie Glabe’s and Rakez Kayed’s studies of an antibody that is specific to small oligomers of different amyloidogenic peptides regardless of their amino acid sequence (see ARF related news story). This surprising study had suggested that the antibody recognizes a shared structural motif on the peculiar oligomeric intermediates of these proteins. At the NBA conference, Glabe, who is at the University of California, Irvine, recapped published data and noted that the list of amyloidogenic proteins known to react with the antibody has since grown to 24.
This September, the scientists further reported that these intermediates all damage synthetic lipid bilayers by a common mechanism that increases the membrane conductance, but that the oligomers do not form pores or specific ion channels in the process (see ARF related news story). At the main Society for Neuroscience conference, Erene Mina, a graduate student in Glabe’s lab (who contributes occasional news summaries to Alzforum), presented a poster describing how treating cultured cells with Aβ oligomers greatly increases calcium influx and disrupts the integrity of the membrane. Soluble oligomers of other amyloidogenic proteins do this, as well, but their respective monomers or fibrils do not. The anti-oligomer antibody reverses the change in conductance. However, much as the scientists had expected to find oligomer pores, they could not. “We see no evidence for discrete conductivity, we see no evidence for open and closed states, and we see no ion specificity. We looked a broad range of inhibitors described as Aβ channel inhibitors, to no effect,” Glabe said at the NBA symposium. “I wanted to see a channel, but this is what we are left with. It’s an urgent issue for us to sort out, and we’ll do it soon.” Other recent work also supports the notion that amyloidogenic oligomers damage the lipid bilayer but not by forming channels (see Green et al., 2004.)
In related news, Glabe also reported these data: immunocytochemistry experiments performed to see whether these oligomers exist in human brain found no immunoreactivity in controls, but a punctate pattern in AD brain. The anti-oligomer antibody detected only a small fraction of total amyloid and did not co-localize with astrocytes or microglia. It occurred largely in extracellular regions that contained plaques but did not overlap with plaques or with diffuse Aβ deposits. Rather, it stained the outer rim of diffuse deposits. The oligomer antibody did not co-localize with neurons, though some intraneuronal staining was apparent. Having solved technical problems with Western blots using this antibody, the lab has now detected the oligomers in soluble extracts of people with MCI and AD, but not controls, Glabe added.
Glabe’s laboratory has begun collaborations with groups studying other diseases. One, with Jeffrey Robbins at Cincinnati Children’s Hospital, shows that human tissue from conditions not previously thought to be amyloid aggregation diseases actually show massive staining with the anti-oligomer antibody. Glabe mentioned forms of idiopathic dilated cardiomyopathy as examples, where oligomeric intermediates might exert their toxicity early and formal fibril aggregation never fully progresses (see Sanbe et al., 2004). The antibody may identify further diseases involving amyloid, Glabe said.
In short, Glabe proffered this working hypothesis: The primary mechanism of all degenerative amyloid diseases lies in the oligomer-induced leakiness of membranes, possibly because oligomers disrupt the way membrane lipids are packed (see also Glabe, 2004).—Gabrielle Strobel.
To be continued Monday, 20 December 2004.
Speakers include:
Randolph Hampton
Paul Muchowski
Ana Maria Cuervo
Ralph Nixon
Cynthia McMurray
Randolph Hampton of University of California, San Diego, introduced the audience to his lab’s work on quality control pathways, which serve to regulate cellular protein levels by proteasome degradation. As an example, Hampton described regulated degradation of HMG-CoA reductase (HMGR), an enzyme in the cholesterol pathway. His lab discovered a natural case of the cell’s use of quality control to set its levels. In this case, in vivo levels of the pathway molecule farnesylpyrophosphate (FPP) determine degradation rates of HMGR. This process itself is modulated by chemical chaperones that stabilize HMGR and decrease access of the requisite E3 ligase, as well as by an FPP derivative that alters the folding state of HMGR to increase access by E3, Hampton said. This work shows that small molecules can affect a protein’s entry into a quality control pathway (see also Shearer et al., 2004).
The underlying mechanism might lead to a therapeutic strategy if drugs could be found that accelerate the recognition of misfolded disease protein by the cells’ quality control, Hampton said. Others in the audience noted that therapies that increase protein turnover generally, such as pathways stimulated by caloric restriction, might prove as useful as protein-specific interventions. That’s in part because the relative contribution of accumulation of specific misfolded proteins and proteasome dysfunction to pathogenesis remains unclear. Moreover, degenerative diseases of aging may well bring with them a broader rise in the level of proteins that need disposal, of which those that researchers know merely represent the tip of the iceberg.
Paul Muchowski tied chaperones into the growing recognition that large protein aggregates may be a lesser culprit in neurodegenerative diseases than early assemblies, however elusive they may be in vivo. He started out by noting that today’s version of the amyloid hypothesis—protein misfolding leads to amyloids, and somewhere along the process the cell dies—applies to many neurodegenerative diseases. Muchowski, who is at the University of Washington, Seattle, asked where in this process chaperones have their greatest effect. He chose to study aggregation of glutamine-expanded (polyQ) huntingtin, which in Huntington disease forms amyloid-like inclusions in the cytoplasm and the nucleus of affected neurons in the striatum and cortex.
Attention began shifting away from inclusions when it turned out that chaperones can prevent neurodegeneration in Drosophila, and also slow neuropathology and motor symptoms in a mouse model of spinocerebellar ataxia, all while leaving inclusions abundantly in place (Kazemi-Esfarjani and Benzer, 2000; Cummings et al., 2001). “This surprised everyone,” Muchowski recalled. It was perplexing, too: If chaperones prevent aggregation, but aggregation visibly occurs in these chaperone-treated models, then how did the chaperones pull off their beneficial effect?
Muchowski hypothesized that mutant huntingtin forms spherical or annular oligomers, and that chaperones prevent their accumulation. The researchers developed a system of controlled expression of the first exon of mutant huntingtin, and added chaperones at various time points in the aggregation process in a five-hour experiment. Spotting the mixture on a grid, they used atomic force microscopy to image and quantify what sorts of species had formed, according to how Brian Caughey and Peter Lansbury had described them for Aβ and a-synuclein. Muchowski’s team saw spherical assemblies form at concentrations well below the threshold for fiber formation; little rings formed at slightly higher concentrations, and fibers appeared above the threshold concentration. When the scientists added the chaperones Hsp40/70 right after turning on mutant ht expression, the oligomers never appeared but instead more fibers formed. Adding the chaperones even an hour into the process had no effect, indicating that chaperones act early in the aggregation process. The data appeared online last month (Wacker et al., 2004).
It’s not clear exactly what the chaperones do, but Muchowski speculates that by simply stabilizing the huntingtin monomer, they cover up certain binding sites needed for the oligomer pathway. One other thing: Muchowski suspects that oligomer formation may represent an offshoot from huntingtin’s major fiber-forming pathway. “It’s not clear at all that oligomers lead to fibers,” he said. This jibes with the observation that reducing oligomer formation actually leads to more fibers, not fewer, and with the finding that chaperones prevent toxicity but not fiber formation in the animal.
The larger question of whether large aggregates are toxic or a protective response by the cell received intense discussion, fueled by Steven Finkbeiner’s recent demonstration that inclusions helped cultured cells loaded with mutant huntingtin survive (see ARF related news story). “The aggregates are a red herring,” said Coleman. “They are there and we can see them. This has been the history with AD ever since Alois Alzheimer described the plaques and tangles. It may turn out that they are not the problem but earlier changes are, and that may be true with other aggregates. They are markers of something else that already happened.”
Therapeutic entry points from this research would appear obvious: Find ways to increase chaperone expression! Indeed, a compound that induces chaperones slows ALS in an SOD mouse model (Kieran et al., 2004), as did a genetic approach in a model of Parkinson’s (Auluck et al., 2002). The hitch, Muchowski said, is that chaperones do more than stabilize aberrant protein monomers. Much like the proteasome, they are prominent players in varied cellular processes. In particular, their role in signal transduction can create a kind of tug of war between neurodegeneration and proliferation. In some cancers, chaperones are overly active, and chaperone inhibition is indeed used in cancer treatment. Therefore, HSP activators would have to be specific and carefully targeted.
One of the many other functions of chaperones that are drawing intense interest in neurodegeneration research is their role in autophagy. By this process the cell degrades waste proteins inside its lysosomes, and it functions in parallel with the ubiquitin-proteasome system. Two complementary talks offered context and new data. First, Ana Maria Cuervo, of Albert Einstein College of Medicine in New York, explained that chaperone-mediated autophagy (CMA) is the most selective of the three known forms of autophagy. The other two major classes of lysosomal protein degradation are macroautophagy, which is inducible and chews up entire organelles, and its constitutively active cousin microautophagy. Both degrade cytosolic components sequestered inside vesicles that are created either de novo in the cytosol (macroautophagy) or pinched off from the lysosome membrane (microautophagy). By contrast, CMA destroys specific cytosolic proteins that translocate across the lysosomal membrane via the lamp2a receptor on the lysosome. Once proteins are inside the lysosome, they are gone within five minutes, but only proteins that carry the “kferq” recognition motif make it there. APP, huntingtin, and α-synuclein all have the kferq motif, and the cytosolic chaperone Hsp70 mediates their recognition and binding to lamp2a, Cuervo said.
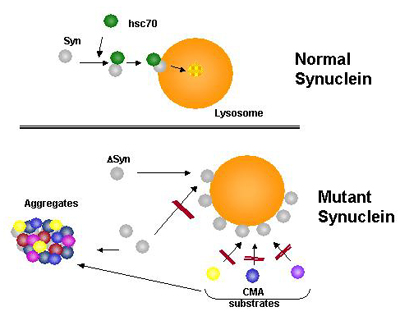
Hypothetical model for the role of altered CMA in familial forms of Parkinson's disease Normal α-synuclein is eliminated from the cytosol through CMA after being recognized by a cytosolic chaperone. Mutant forms of α-synuclein are targeted to the lysosomal membrane and bind to the receptor but do not translocate. The high affinity of the mutant protein for the lysosomal receptor results in blockage of the translocation complex for other CMA substrates, that are likely to also accumulate in the cytosol of the affected cells.
Cuervo first outlined a series of experiments suggesting that CMA participates in the removal of damaged proteins. It responds to starvation, toxins, and oxidative stress in a variety of different cell types. Lysosomes isolated from cultured cells put under mild oxidative stress, or from mice injected with the toxin paraquat, both contained increased numbers of oxidized cytosolic proteins. Oxidized proteins translocate better into lysosomes, perhaps because they partially unfold, and CMA responds to paraquat stress by boosting the lysosomes’ capacity to import oxidized proteins.
With aging, CMA activity decreases while levels of oxidized cytosolic proteins increase. Cuervo suggested that the former contributes to the latter, and that CMA is where aging cells became particularly vulnerable to oxidative and toxic stress. To begin testing this assumption, the researchers interfered with lamp2a expression in fibroblasts. This led to oxidized proteins’ being stuck outside lysosomes and impaired the cells’ ability to survive conditions of oxidative and toxic stress.
Cuervo’s lab then set out on a foray into neurodegeneration by studying α-synuclein’s behavior on lysosomes (see ARF related news story). In a nutshell, the scientists show that both of the two α-synuclein mutations known to cause Parkinson disease bind more tightly than wild-type α-synuclein to the lysosomal lamp2a receptor, but they never enter the vesicles inside. In this way, mutant α-synuclein sticks around, and lysosomal receptors remain loaded so that other substrates cannot enter for their degradation, either. “As a consequence of CMA blockage, damaged proteins accumulate in the cytosol, and the cell loses its ability to respond to stress,” Cuervo said.
Cuervo’s work offers opportunities for collaboration with other labs working on neurodegeneration, particularly by way of cross-breeding her new line of mice. Cuervo and colleagues made a strain of mice, still young, in whom they can induce overexpression of lamp2a at will. They plan to activate lamp2a when the mice age and ask whether putting more receptors into lysosomes can prevent the age-related decrease in CMA, or even reverse the age-related accumulation of damaged proteins. Other future work will be directed at studying differences between oligomeric forms of α-synuclein and their effect on the lysosomal membrane (see Yang et al., 1008). Cuervo also plans to investigate the major AD risk factor ApoE, whose isoforms are known to differ in their lysosomal degradation (see Mahley section of ARF related news story, Yi et al., 2002).
Cuervo emphasized that researchers should not focus on one cellular degradation system exclusively. In her experience, experiments manipulating CMA lead to changes in other degradation systems, as well, as these systems crosstalk. For example, chronically blocking CMA in cultured cells leads, paradoxically, to an increase in overall degradation. That’s because macroautophagy kicks in to compensate, and proteasomal degradation also responded by changing the subunit composition of the proteasome. “Proteasomal degradation clearly crosstalks with lysosomal degradation, and this is why there is so much confusion in the proteasome field. This crosstalk is very important. We need to consider all these pathways and learn how they interact,” Cuervo said.
Ralph Nixon began his talk by seconding Cuervo’s appeal that scientists studying a given cellular protein degradation system always keep an eye on the others. He then focused on broader changes in autophagy in AD. In this disease, the lysosomal system becomes mobilized early on as a protective response, but it fails to pull all the way through to protein degradation and instead gets backed up somewhere in the process. This leads to pathologies that deserve more study, Nixon said. Bob Terry described accumulating lysosomes in AD (Suzuki and Terry, 1967), but this observation has been forgotten. “Now we realize how prominent this is,” Nixon said.
Autophagy begins with the formation of membrane sacs called autophagosomes, which then fuse with late endosomes or lysosomes for degradation of their contents. In normal brain this process is barely detectable, but most neurons in AD show evidence of it and do so with a massive pathology, Nixon said. Using antibodies that detect induction of autophagy, Nixon’s group saw an upregulation of the process in dendrites of hippocampus and cortex of PS/APP-transgenic mice that begins at nine weeks, before the mice deposit amyloid. By nine months, when amyloid deposition is in full swing, the autophagy antibody labels the dystrophic neurites known to course through and around plaques. In fact, autophagic vacuoles (AVs) become the predominant organelle in dystrophic neurites.
What stimulates this? Experimentally, sublethal Aβ levels and oxidative stress both induce autophagy in cultured neurons, Nixon said. What’s more, he believes that the presenilin protein may have a normal function in autophagy. Nixon’s collaborator Anne Cataldo, now at McLean Hospital in Belmont, found that FAD presenilin mutations cause a more severe disturbance of lysosome function than is seen in sporadic AD, and APP/PS double-transgenic mice have more pronounced lysosomal pathology than do mice transgenic for APP alone, (Cataldo et al., 2004). Fibroblasts from people with PS1 mutations showed a proliferation of AVs and kept accumulating them, unable to complete protein degradation, Nixon said. Blastocysts from PS1/2 double-knockouts failed to induce autophagy, leading Nixon to suggest that presenilin is required for autophagy. This suggests a presenilin function that is independent of APP cleavage but would be required for the clearance of amyloid through the lysosomal pathway, not to mention the many other proteins and organelles turned over by autophagy. It is unclear which presenilin substrate is required for autophagy to work properly. Since in FAD, autophagosomes accumulate and, in PS-null blastocysts, grow to “monster” proportions, Nixon said, it is likely to be one that comes into play later, perhaps during lysosome fusion.
Nixon also noted evidence that the components required for Aβ production are enriched in isolated AVs (Yu et al., 2004), suggesting that these autophagic organelles might continuously produce intraneuronal Aβ as they accumulate. “Peripherally, we have evidence that autophagy is a route for Aβ production,” Nixon said. Tau pathology also stimulates autophagocytosis, but Nixon questioned whether this process is an early manifestation of AD. Rather, tau dysfunction may tie autophagy and axonal transport into a broader picture.
What, then, is the earliest insult that promotes Aβ generation and, in turn, autophagy? No consensus pathway exists, but Nixon pointed to dysfunction in neuronal endocytosis as his favorite suspect. In this scenario, early endosomes do not mature and instead enlarge (Cataldo et al., 2004). Signaling endosomes do not travel to the nucleus to transmit protective growth factor signals, tying axonal transport into the picture (see also ARF related conference story).
Picking up the baton where Nixon had left off, Cynthia McMurray, in a rapid-fire presentation on Huntington disease (HD), concurred that defects in endocytosis are the earliest sign of trouble her experiments are picking up. To set the stage, McMurray, who is at the Mayo Clinic in Rochester, Minnesota, first recapped that while some molecular processes of HD overlap with those in other neurodegenerative diseases, HD's genetics are unusual. The defect is not a missense mutation or deletion, but an expansion of CAG triplet repeats, and the length of the expansion determines age of onset. Curiously, the length of the expansion grows from generation to generation, such that the disease can begin at age 50 in a grandfather, 26 in his daughter, and six months in her child, reflecting a lengthening of the repeats from 92 in the first generation to over 200 in the third.
A thumbtack version of McMurray’s view of Huntington’s goes like this: Toxicity arises from an oxidation cycle that couples htt protein and DNA. Mutant huntingtin first causes defects in endosome and axonal trafficking. This impairs mitochondria, which then secrete toxic radicals. The radicals modify DNA, and subsequent attempts to repair the damage create DNA breaks, which lead to somatic expansion of the CAG repeat. In this cycle, the trafficking defect sets off toxicity, and the oxidative/DNA damage cycle continues throughout life. Inclusions form as a consequence of the critical toxic events, McMurray contends.
McMurray’s experiments use mice and cultured striatal neurons expressing full-length mutant human huntingtin (mhtt). “All my work uses the endogenous whole protein. I don’t think expressing just an exon or two gives you the right answer,” McMurray said. Early on in her studies, the cultured neurons retracted their axons within two days of expressing mhtt because traffic along microtubules faltered. A closer look revealed that the first step in this defective process lay in little flask-shaped indentations of the cell membrane called caveolae that were not being properly endocytosed and trafficked anymore. Found in most tissues, caveolae constitute a system of endocytic transport vesicles that operates alongside the better-known clathrin-coated pits. They remain enigmatic but are of particular intrigue in neurodegenerative disease because their constituent proteins, the caveolins, also serve as scaffolding hubs that recruit signaling molecules to the caveolae. This is important for the function of signaling endosomes and has been implicated in Parkinson disease (Hashimoto et al., 2003).
McMurray found that caveolin protein interacts strongly with the mutant htt, but not with normal htt. This weakens caveolin’s required interaction with cholesterol. Caveolae from the mutant htt-expressing neurons were unable to efflux cholesterol from the neurons, and cholesterol piled up inside them. This observation held in vivo, as the scientists correlated large intraneuronal increases in cholesterol with the appearance of a clasping motor phenotype in HD mice. In culture, turning mutant htt expression on induced a cholesterol increase, and silencing the gene restored proper trafficking of caveolae and brought cholesterol levels down, McMurray said. She suspects that this cholesterol accumulation compromises the nuclear membrane in such a way that huntingtin—otherwise too large a protein to enter the nucleus under normal conditions—can now enter. Even so, McMurray attributes all the changes leading up to this nuclear translocation to htt in the cytoplasm and contended that nuclear htt has little to do with the primary pathophysiology of HD (see also ARF related news story). “I really think nuclear aggregation is a late event,” McMurray said.
Caveolar membranes are of the lipid raft type, and McMurray’s team is now studying how their lipid content changes. This finding has echoes in other neurodegenerative diseases where altered cholesterol homeostasis is a problem. For example, Niemann-Pick syndrome is primarily a disease of cholesterol storage in lysosomes, and ApoE, which receives cholesterol from caveolae, is a risk factor in AD.
The early endocytic problems then grow into broader trafficking defects along microtubules. Mitochondria in htt-mutant mice stop moving even before neurological symptoms develop, probably because mutant htt sequesters wild-type htt along with motor proteins and other trafficking components. This work, recently published (Trushina et al., 2004), confirms in mammals earlier work done in invertebrates (see ARF related news story).
A second line of investigation traces what happens to the cell after damaged mitochondria begin spewing out hydroxyl radicals. McMurray said she believed that the obvious consequence—protein and lipid oxidation—wreaks less havoc than what goes on at the DNA. There, hydroxyl radicals launch a process that leads to a cycle of lengthening of the repeat size and aggravated htt toxicity in a given person as (s)he ages, McMurray contends.
The growth of the htt expansion can be shown in HD mice and in humans. To test the idea that hydroxyl radicals instigate it, McMurray and colleagues isolated embryonic cells from their HD mice and treated them with hydrogen peroxide. This prompted the existing triplet repeats to lengthen with time. Further studies detailed a process by which the DNA repair enzyme 8-oxoG-DNA glycosylase (OGG1) cuts out oxidized bases from the DNA, creating a strand break. In their experiments, this strand lifted off the DNA duplex and formed a stable loop with itself, leaving behind a single-stranded gap that DNA polymerases subsequently filled in. Also left behind was a longer expansion in the aberrant loop. It gets transcribed to yield a more toxic htt protein, and it provides more spaces for oxidation and further elongation with age. This process was more active in brain than in liver, McMurray noted. To test this model, the scientists bred mice lacking the OGG1 enzyme with mice expressing mutant htt, and the crosses indeed do not show repeat expansion with age. Preliminary data suggest that the crosses develop a milder form of disease, as well, McMurray said.—Gabrielle Strobel.
No Available Comments
Comments
No Available Comments
Make a Comment
To make a comment you must login or register.