CONFERENCE COVERAGE SERIES
Kuopio Alzheimer Symposium (9th) and Nordic Memory Clinic Conference (3rd)
Kuopio, Finland
23 – 25 August 2022

CONFERENCE COVERAGE SERIES
Kuopio, Finland
23 – 25 August 2022
The 9th Kuopio Alzheimer Symposium and 3rd Nordic Memory Clinic Conference took place jointly on August 23-25 at the Kuopio Music Center, a modern performing arts and conference center in this Finnish city just short of the Arctic Circle. Co-organized by Mikko Hiltunen, University of Eastern Finland, Kuopio, this northernmost dementia conference drew about 300 people from Scandinavia, elsewhere in Europe, and some from Asia and the Americas, too. On the agenda were diverse topics ranging from genetics and biomarkers to prevention and therapies. One take-home message from the meeting: People who live past 100 may enjoy built-in resilience to brain disorders such as dementia.
That’s one conclusion from the 100-plus Study, an observational cohort being followed by Henne Holstege and her colleagues at Amsterdam University Medical Center in the Netherlands. Holstege reported that while some centenarians do accumulate pathologies associated with aging and neurodegeneration—think cardiovascular damage and proteopathies—these don’t necessarily lead to cognitive decline. The reason? The very old are protected by their genetics.
For example, cognitively healthy centenarians have a much lower genetic risk for Alzheimer’s disease, Holstege said, attributing some of that to a variant in the gene for phospholipase C-γ2. An analysis of nine families who carry the PLC2G P522R variant suggests that it boosts the immune system. “While the effects are small, the data point to PLCG2 as a potential therapeutic target,” said Holstege.
“This work on centenarians is super important,” said Henrik Zetterberg, University of Gothenburg, Sweden, who attended the Kuopio meeting. “It’s almost like their amyloid pathology is inert, or surrounded by something that shields it,” he speculated.
Holstege began the study in 2013, inspired by Hendrikje van Andel-Schipper, who, at 115, was the oldest woman alive when she died in 2005. Van Andel-Schipper had shown no signs of dementia. “She demonstrated that cognitive decline is not inevitable in people who live to very old age,” said Holstege. Van Andel-Schipper’s mother had lived to 100 and she, too, had her full faculties. Does the van Andel-Schipper genome—and that of other cognitively healthy centenarians—hold clues to dementia prevention or treatment? Holstege started the 100-plus Study to find out.
In Kuopio, Holstege reviewed some of the latest data from the project. She initially aimed to recruit 500 cognitively healthy centenarians. As of June 2021, 406 had signed up. Their average age when they joined was 101; the oldest is now 107. Seventy percent are women; 43 percent still live independently. About 30 percent have agreed to donate their brains after death and, to date, 95 of them have passed away. Holstege showed neuropathology findings from 85 of those.

100-Plus Study. The project scours the blood and tissues of healthy centenarians for clues to dementia prevention and treatment. [Courtesy Holstege lab, www.holstegelab.eu.]
At autopsy, some of these centenarians were found to have had pathologies typical of people with AD. Ph.D student Andrea Ganz from Holstege’s lab and Meng Zhang, from Marcel Reinder’s lab at Technical University Delft, The Netherlands, saw that many had been in stage 2 or 3 amyloidosis when they died, as judged by NIA criteria, and had accumulated stage 2 or even stage 3 neuritic plaques per CERAD scores. All were in at least Braak stage I for neurofibrillary tangles, though the majority were at stage III or higher. The brains weighed about as much as those from people who had had AD dementia, but neither plaques nor tangles correlated with MMSE score.
The same was true for a host of other age-related pathologies, including cerebral amyloid angiopathy, TDP-43 proteinopathy, Lewy bodies, hippocampal sclerosis, granulovacuolar degeneration, atherosclerosis, and vascular infarcts. Many centenarians had at least one of these. Across all of these pathologies, increased levels in the postmortem brain generally came with lower cognitive scores before death, but the associations were weak. Of all neuropathological substrates tested, tangles correlated most robustly with lower performance, but these healthy centenarians seemed surprisingly resistant even to the effects of high levels of tangles, Holstege said. A manuscript on this data was uploaded to medRXiv on August 30 (Zhang et al., 2022).
So what is going on in these brains? To investigate this question at a systems biology level, Holstege collaborated with Guus Smit in Amsterdam UMC, who studies bulk brain proteomes. The scientists compared the centenarians' proteomes to those from 50- to 95-year-old people with AD and to 50- to 90-year-old healthy controls. They found that, while concentrations of about two dozen proteins fall with age, in centenarians, these protein levels were higher than expected for their age. For four other proteins that normally tick up with age, levels remained steady in the centenarians. The proteins that bucked these trends included those involved in microtubule and intermediate filament biology, myelination, the immune system, basic metabolism, and protein transport. “In a nutshell, these centenarians have younger-looking brains,” said Holstege.
What bestows the vim and vigor? Holstege believes genetics holds the key. Based on 83 known genetic risk variants for AD, Niccoló Tesi, also at Amsterdam UMC, found that centenarians have a much lower polygenic risk score for this disease than do people with AD. Indeed, in a previous, smaller analysis of about two dozen AD gene variants, people with AD were likelier than cognitively healthy centenarians to have risk variants in APOE and TREM2, but less likely to have protective variants in other genes, including PLCG2 (Tesi et al., 2019). “There is a depletion of risk variants and an enrichment of protective variants versus age-matched controls,” said Holstege.
Scientists had previously found the P522R protective variant in PLCG2, and that it may boost immune function and indeed longevity (Aug 2017 conference news; Sep 2020 news). Holstege learned that it also protects against frontotemporal dementia and dementia with Lewy bodies, and that people who live past 90 are much likelier to carry this variant (May 2019 news).
To explore more deeply how the P522R protective variant in PLCG2 might function, Holstege and colleagues identified nine families—beyond the centenarian cohort—who carry it. Annieck Diks works with Holstege’s collaborator Jacques van Dongen in Leiden University Medical Center. She found that 33 carriers of the variant seemed to have blood cells counts that resembled those of younger people. Their B cells seemed more sensitive to stimuli, more ready to phosphorylate PLCG2 and release intracellular calcium. Their myeloid cells made more reactive oxygen species and phagocytosed more avidly than did cells from siblings who did not carry this particular variant. Diks uploaded a paper on this data on the Research Square preprint server (Diks et al., 2022).
Researchers in Kuopio called findings outstanding. Holstege said these outcomes were expected given similar findings in mouse studies. To her knowledge, this was the first data on freshly isolated human peripheral immune cells. “Admittedly, these are peripheral cells, but for now we think this is a good model for what might go on in the brain,” she said.
The Hiltunen lab at UEF studies how the PLCG2 variant affects brain cells as part of a personalized medicine project for newly discovered microglia-associated genetic variants in AD. For this project, called PMG-AD, Hiltunen collaborates with four other investigators: Christian Haas, Ludwig-Maximilians University, Munich; Jari Koistinaho, University of Helsinki; Jean-Charles Lambert, Institut Pasteur de Lille, France; and Stefan Lichtenthaler, Technical University of Munich.
PMG-AD identifies carriers of the PLCG2 P522R variant, among others, in several cohorts, including the Alzheimer Disease European Sequencing consortium, the European Alzheimer’s Data Bank, Alzheimer's Disease Genetics Research Unit, and FINGER—the Finnish multimodal intervention trial. The researchers will probe microglia derived from blood monocytes and iPSCs for functional effects of the variants. The goal is to correlate those with transcriptomic, proteomic, and amyloid and microglial PET data in search of protective or disease markers or drug targets.
Hiltunen also collaborates with Holstege to characterize the molecular and cellular mechanisms that protect her study's centenarians. “I strongly believe that the pool of protective genetic variants dictates a lot in these individuals,” he told Alzforum. “The PLCG2 variant may even override the adverse effects of APOE4.”
“All in all, studies done in this 100+ cohort provide seminal information on key determinants of resilience and longevity, including genetic variants. This may provide specific targets to tackle in Alzheimer's and other neurodegenerative diseases,” he said.—Tom Fagan
No Available Further Reading
Few scientists doubt that backed-up clearance of detritus from the brain correlates with accumulation of amyloid plaques and neurofibrillary tangles. But which comes first? Data presented at the 9th Kuopio Alzheimer Symposium suggests that, at least in some cases, the plumbing might be the problem. Per Kristian Eide, University of Oslo, Norway, reported that the cerebrospinal fluid does not drain properly in people who have normal-pressure hydrocephalus. Is there an AD connection, you may ask? In people with this brain condition, the incidence of dementia, and amyloid and tangle pathology, far exceeds that of the general population. Eide’s data support the idea that sluggish drainage of CSF may cause AD pathology.
A rare condition, idiopathic normal-pressure hydrocephalus typically manifests as a troika of symptoms, that is, a gradual loss of balance, bladder control, and cognition (Williams et al., 2016). It primarily affects older people, and about 6 percent of octogenarians are estimated to have the disorder (Jaraj et al., 2014; Iseki et al., 2022). Brain scans show that these people have enlarged ventricles filled with CSF.
On autopsy or biopsy, a small majority of people with NPH and dementia also have amyloid plaques and neurofibrillary tangles, the hallmarks of AD pathology (Cabral et al., 2011; Koivisto et al., 2016). In many cases, the tangles can be sparse, with little evidence of neurodegeneration, suggesting that there might be an opportunity to intervene early in their AD progression (Libard and Alafuzoff, 2019).
Indeed, some people who have a shunt surgically implanted in their brain to drain the CSF make remarkable recoveries and their cognition improves (Adams et al., 1965; McGirt et al., 2005; Liu et al., 2016). However, this is far from true for everyone. Researchers in Ville Leinonen’s lab at Kuopio University Hospital reported that four years after this procedure, 80 percent of people had cognitive decline and 46 percent had either Alzheimer's or vascular dementia (Koivisto et al., 2013).

Trace The Fluid. Serial MRI scans show that after injection into a healthy person's cerebrospinal fluid, the MRI contrast agent gadobutrol gradually spreads throughout his or her brain, depicting a normal glymphatic system. [Courtesy of Per Kristian Eide.]
What causes NPH? In some cases, called “noncommunicating NPH,” it can be a blockage that restricts CSF flow between the brain's ventricles. In “communicating NPH,” the obstruction occurs after CSF leaves the ventricles. In many cases the cause is unknown.
To study the flow of CSF in idiopathic NPH, Eide and colleagues tested the MRI contrast agent gadobutrol. This gadolinium compound is used in animals to image the glymphatic system, by which interstitial fluid moves through the brain and exchanges with the cerebrospinal fluid (Aug 2012 news; Mar 2013 news).
In Kuopio, Eide showed that when the tracer is injected into the spine of a healthy volunteer, it slowly spreads into the parenchyma of the brain (Ringstad et al., 2018). This distribution peaks up to a day later, Eide said (see image above). Because the resolution of MRI is only around 1 mm, it can't depict the exact path the tracer takes.
Even so, Eide showed that it flows beside arteries, without penetrating them. This is in keeping with the idea that fluid in the brain spreads by the paravascular glymph system, whereby CSF flows into the parenchyma along arteries, and drains out of it along veins (Aug 2012 news). In keeping with this, the strongest spread of gadobutrol into the parenchyma occurred around major arteries. Eide thinks that the pulsing of the arteries helps to gently push the fluid along.

Brain Drain? The MRI tracer gadobutrol makes its way from the CSF (light blue) to the parasagittal dura (yellow), which harbors lymph vessels that drain the brain (not shown). [Courtesy of Ringstad and Eide, 2020.]
Does this paravascular transport system change in iNPH? Geir Ringstad at Oslo University Hospital compared gadobutrol clearance among eight healthy controls and 15 people with iNPH (Ringstad et al., 2017). Ringstad found that, in iNPH, the tracer is slow to enter and exit the Sylvan fissure. This major sulcus separates the frontal and parietal lobes from the temporal lobe. It is a main point of entry for gadobutrol into the parenchyma from the CSF. What’s more, once the agent was in the parenchyma, it cleared more slowly than it did in healthy controls.
Following these findings, Eide and colleagues modeled how gadobutrol drains from the brain into the blood. First, some background. Scientists know that the meninges and dural membranes of mouse and human brains contain a set of lymph vessels that likely sieve interstitial fluid pushed through the brain by the glymph system (Aspelund et al., 2015; Louveau et al., 2015; Oct 2017 news). Eide and colleagues had found that gadobutrol squeezes from the interstitial fluid into the parasagittal dura at the top of a person's brain, which houses some of these lymph vessels (Ringstad and Eide, 2020). Eide believes that the parasagittal dura serves as a bridge between the CSF and the lymph, and ultimately, the blood.
Now for the blood modeling. Markus Herberg Hovd and colleagues in Eide’s lab injected gadobutrol into the CSF of 161 volunteers, then measured it in blood samples taken at intervals for up to 48 hours (Hovd et al., 2022). Of these people, 28 were healthy controls, 63 had iNPH, and 70 had another form of hydrocephalus or CSF disorder. At the Kuopio conference, Eide showed how Hovd used the blood data to model the pharmacokinetics of brain clearance, arriving at a formula that closely matched the empirical data.
He then plugged blood sample data for each volunteer into the formula to determine clearance pharmacodynamics of each volunteer (see graphs below), including how long it took half of the tracer to clear the CSF, time to maximum concentration in the blood (Tmax), the peak concentration in blood (Cmax), and the lag time before the tracer appeared in the blood (Tlag).
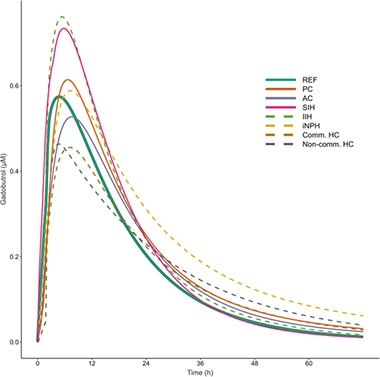
Clearance Problems. Modeling CSF clearance by tracking the appearance of gadobutrol in the blood suggests that people with iNPH (yellow dashes) take longer to clear solutes from the brain than do healthy controls (solid green). Pharmacodynamics also changed in people with other forms of NPH (PC, pineal cyst; AC, arachnoid cyst; SIH, spontaneous intracranial hypotension; IIH, idiopathic intracranial hypertension; Comm HC, communication hydrocephalus; Non-comm HC, non-communicating hydrocephalus). [Courtesy of Hovd et al., 2022.]
The first thing Hovd noticed was that the model indicated that the volunteers, even the controls, were markedly different from one another on all those parameters. “This considerable variability really surprised us,” said Eide. Still, differences emerged between the groups. For example, in 11 people who had communicating hydrocephalus, the Cmax was about 30 percent lower than in controls. In 13 people who had a pineal cyst, Tlag was more than twice as long. That said, people with iNPH had the most dramatic changes. Both their Tlag and Tmax were longer than in the reference group, meaning the tracer hung around longer in their brains. Ultimately, gadobutrol reached higher concentrations in the blood of people with iNPH, but Eide thinks this happened because their kidneys were affected, too, and cleared the tracer more slowly.
It remains to be seen whether this abnormal CSF drainage in iNPH contributes to AD pathology, as has been shown in animal models. Next, Eide plans to measure gadobutrol clearance in people with mild cognitive impairment.—Tom Fagan.
No Available Comments
Scientists now have a slew of biomarkers, both fluid assays and PET ligands, that distinguish Alzheimer’s from other neurodegenerative diseases, such as frontotemporal dementia, Parkinson’s, and dementia with Lewy bodies (DLB). But in the great many cases where there is mixed pathology, the overlaps are messy and differential diagnosis often gets stuck. Could synaptic proteins ride to the rescue? At the 9th Kuopio Alzheimer’s Symposium held August 23-25 in this city in Eastern Finland, VAMP2 emerged as a candidate to distinguish pure DLB from DLB with evidence of concomitant AD pathology.
Olivia Belbin, IIB Sant Pau, Barcelona, Spain, reported that levels of this synaptic protein go down in the former, but up in the latter. “This suggests that different processes are at play in these diseases,” she said.
Henrik Zetterberg, University of Gothenburg, Sweden, was intrigued. “I know of no other marker that behaves like this,” he said, emphasizing that the findings need to be replicated. “If this is confirmed, then we really need to dig into cell-based mechanisms and other studies to find out what is going on with this marker,” he said.
VAMP2 stands for vesicle-associated membrane protein 2. It forms SNARE complexes with its fellow synaptic proteins SNAP-25 and Syntaxyn-1. These complexes help deliver synaptic vesicles to the presynapse and fuse them with the membrane there. Neurons can assemble and disassemble SNAREs thousands of times per minute. Scientists suspect SNAREs might prove to be sensors of change in synaptic health. Hence, they are studying whether SNARE levels in the parenchyma or CSF correlate with neurodegenerative pathologies in general, and with α-synucleinopathies in particular (see Cervantes-González and Belbin, 2022, for a review).
Older work from Nobel laureate Tom Sudhof’s lab indicates that α-synuclein promotes formation of SNARE complexes, a function that might be compromised by synuclein oligomers or aggregates (Burré et al., 2010). Last year, researchers in Italy reported that levels of VAMP2 in neuron-derived extracellular vesicles fell in people who had Parkinson’s disease (Agliardi et al., 2021). Those same scientists reported this year that people diagnosed with mixed dementia express more of the VAMP2 gene in their brains than do people who are cognitively normal or who have AD, and that these mixed dementia folk are more likely to have a 26 base pair deletion in an intergenic region of the gene (Costa et al., 2022). All told, interest in VAMP2 has recently blossomed.
Belbin was searching for markers that identify heterogeneities in neurodegenerative diseases. She noted that a majority of Alzheimer’s cases also have some other neuropathology, for example Lewy bodies, which comprise mostly α-synuclein aggregates. To find changes in VAMP2 in the CSF of people with AD and related disorders, Belbin developed a highly sensitive “homebrew” SIMOA immunoassay. It uses a rabbit monoclonal antibody from the company Cell Signaling Technology to capture the protein, plus a high-affinity mouse monoclonal developed by ADx NeuroSciences for detection. The assay's lower limit of quantification is 4.1 pg/mL, said Belbin.
Armed with this tool, Belbin measured CSF VAMP2 in samples from the Sant Pau Initiative for Neurodegeneration (Alcolea et al., 2019). Begun in 2011, SPIN enrolls healthy controls, people with subjective memory complaints and mild cognitive impairment, and people who have been diagnosed with AD, DLB, frontotemporal dementia, or Down’s syndrome.
In Kuopio, Belbin showed data from 63 healthy controls, 114 people with AD, 19 with pure DLB, and 28 with both DLB and AD pathology. On average, the latter had more VAMP2 in their CSF than did controls, but curiously, people with pure DLB had less VAMP2 than controls (see image below). In this way, the assay was able to distinguish people with pure DLB from those with DLB+AD with an AUC of 0.80—the AUC is a measure of sensitivity and specificity, with a value of 1.0 indicating a perfect assay.

Differential Diagnosis? These violin plots suggest that people with pure DLB have less VAMP2 in their CSF on average, than people with AD, or DLB+AD (left). In early AD, however, VAMP2 reaches a low point. [Courtesy of Olivia Belbin.]
Why the apparent difference between DLB and DLB+AD? Belbin thinks that in pure DLB, VAMP2 decreases in the CSF because it is sequestered by aggregates of α-synuclein. A recent mass spectrometry study spotted VAMP2 among a cadre of synaptic proteins that were embedded in Lewy bodies purified from people with DLB (McCormack et al., 2019).
But why would VAMP2 increase in the CSF of AD patients? Belbin thinks this might reflect damage to synapses and/or neurodegeneration. In fact, when she looked across different stages of AD, she found less VAMP2 in the CSF of people who were amyloid-positive and tau-negative than in CSF of controls, but more VAMP2 in CSF of amyloid-positive/tau-positive people, i.e., those further along in their pathogenesis. The data suggest that as AD develops, CSF VAMP2 first drops, then rises. This rise correlated tightly with levels of phospho-tau in the CSF, Belbin said. Belbin found a similar nonlinear progression in Down’s syndrome (Lleó et al., 2021).
Zetterberg called the data fascinating. “There are interplays here between α-synuclein and AD pathology, and you have to ask what role amyloid plays,” he said. Synapses contain large amounts of α-synuclein. "If synapses are exposed to amyloid, then maybe both together cause harm in a way that affects this synaptic marker,” he suggested. In fact, fluorescence resonance spectroscopy suggests that amyloid might disrupt interactions between VAMP2 and SNAP-25 (Sharda et al., 2020).
Belbin also showed that, in CSF samples from the University of Pennsylvania FTD Center and the UPenn Alzheimer Disease Research Center, VAMP2 was elevated in AD but unchanged in people who had either FTLD-tau or FTLD-TDP-43 (Cervantes González et al., 2022). “This again speaks to a role for amyloid,” said Zetterberg.

More Violins. On average, people with AD have more VAMP2 in their CSF than do healthy controls or people with FTD-tau (left) or FTD-TDP-43 (second left). [Courtesy of Olivia Belbin.]
Still, Belbin thinks VAMP2 may help clinicians who treat FTD tease out whether the patient in front of them suffers from FTLD-TDP-43 or from FTLD-tau. In a separate, mass spectrometry-based, shotgun proteomics study, she identified more than 4,000 proteins in human CSF, of which more than 250 were synaptic (Lleó et al., 2019). Using a targeted mass-spectrometry assay to quantify a panel of nine of these synaptic proteins in CSF samples from UPenn, Belbin reported in Kuopio that reduced levels of calsyntenin-1 and neurexin-2a in CSF, taken while volunteers were alive, correlated with their postmortem levels of TDP-43 pathology, but not with tangles.
Belbin showed that a multi-marker proteomic panel made up of calsyntenin-1, the AMPA receptor subunit GluA4, and VAMP-2 distinguished FTLD-TDP-43 from FTLD-tau with an AUC of 0.83. This panel correlated with MMSE scores, whereas individually, none of its component markers correlated with cognition. Belbin thinks this panel might help trialists pick suitable participants for FTD clinical trials of drugs targeting tau or TDP-43, respectively. It could also serve as a surrogate for cognitive performance, she said.—Tom Fagan.
No Available Comments
No Available Further Reading
Comments
No Available Comments
Make a Comment
To make a comment you must login or register.