CONFERENCE COVERAGE SERIES
Human Amyloid Imaging 2009
Seattle, WA, U.S.A.
24 April 2009
CONFERENCE COVERAGE SERIES
Seattle, WA, U.S.A.
24 April 2009
Slowly but surely, Alzheimer disease researchers are coming to grips with the possibility that some experimental therapies could be failing because they have been tested in people whose disease is too advanced. The field’s focus is therefore shifting toward earlier diagnosis, even prevention. Reflecting the new emphasis, this year’s Human Amyloid Imaging (HAI) meeting drew some 150 researchers to Seattle on 24 April to share and discuss the latest in brain imaging, which will be crucial for identifying at-risk individuals and helping them resist impending AD.
A growing literature documents elevated brain amyloid in a substantial proportion of seniors who appear cognitively normal. This finding has stirred up new questions—not the least of which is whether this amyloid foretells future AD. That issue remains to be clearly resolved. However, ask any number of researchers at the HAI meeting, and chances are they’ll reckon that having a head full of amyloid is worrisome. “There was consensus that among normal people, amyloid is associated with changes in the brain,” said Reisa Sperling of Brigham and Women's Hospital in Boston, Massachusetts, in a conversation with this reporter during the poster session. “That might be very valuable in identifying individuals who will get preventive treatment. If we can identify people who are going to get AD a decade later, we have a window to treat people before they get symptoms,” Sperling noted.
To address that “if,” HAI’s opening session explored the relationship between amyloid deposition and functional changes in the brain. Each can be measured by positron emission tomography (PET)—the former with radiolabeled amyloid tracers, the latter using fluorodeoxyglucose (FDG) metabolism. Previous live brain imaging with the PET tracer Pittsburgh Compound-B (PIB) has revealed elevated Aβ in 10 to 30 percent of cognitively normal elderly, and has shown that amyloid load tracks longitudinally with whole brain atrophy. Elizabeth Mormino, a Ph.D. student in Bill Jagust’s lab at the University of California, Berkeley, addressed whether high amyloid deposition also coincides with reduced glucose metabolism, i.e., portends a loss of brain function. In her study, normal seniors and AD patients received magnetic resonance imaging (MRI) to track brain atrophy, FDG-PET to measure glucose metabolism, and PIB-PET to detect amyloid load.
Based on a published method for defining cut-offs (Aizenstein et al., 2008), all AD patients in Mormino’s group were classified as “high PIB,” as were 11 of 40 normal participants. Among the remaining healthy seniors, 21 came up as having “low PIB,” and the rest fell into an intermediate zone. The subjects were also grouped according to FDG metabolism. Within the cognitively normal group, high PIB was associated with reduced glucose metabolism, albeit with FDG-PET patterns less pronounced than in demented populations. Among those with high PIB, lower glucose metabolism also correlated with worse episodic memory. This trend did not hold for the people with low PIB. All told, the data suggest that elevated PIB uptake in normal seniors may suggest preclinical AD, Mormino said.
Ann Cohen, a postdoctoral fellow in Bill Klunk’s group at the University of Pittsburgh, Pennsylvania, also reported a link between amyloid load (PIB-PET) and cerebral metabolism (FDG-PET). In her study of 51 healthy seniors, 38 had high PIB uptake and 13 fell into the low-PIB group. Cohen used software that establishes cut-offs for “abnormal” and “normal” metabolism through automated iterative analysis of FDG-PET scans. By this method, Cohen further subdivided PIB groups according to glucose metabolism status. On her poster, Cohen reported that PIB-positive elderly were six times more likely to have abnormal metabolic patterns than were PIB-negative people. On the flip side, those with disrupted glucose metabolism were four times more likely to have elevated brain amyloid. However, a handful of participants had discordant PIB and FDG profiles, i.e., high PIB with normal FDG-PET, or low PIB with abnormal FDG-PET. This suggests that the correlation between the two techniques is not airtight. “People are realizing more and more that you need a multi-modal approach,” Cohen said.
Inconvenient and expensive as it is, this conclusion also emerged in a talk by Gil Rabinovici of the University of California, San Francisco. Rabinovici compared the value of PIB- and FDG-PET in distinguishing clinically diagnosed AD and frontotemporal lobar degeneration (FTLD). He chose FTLD because it is a non-Aβ dementia whose patients tend to be younger, which makes age-related amyloid less of a concern. Using either PIB or FDG correctly diagnosed about 80 percent of the patients (40 AD, 36 FTLD), but each method misclassified a different set of people. Hence, though the techniques showed high diagnostic sensitivity and specificity, they agreed with each other moderately at best, suggesting to Rabinovici that they are “far from redundant and are providing us with complementary information.”
In a similar vein, Shizuo Hatashita of Shonan-Atsugi Hospital and Clinic in Kanagawa, Japan, presented data suggesting that while brain amyloid load may indeed predict future development of AD, it seems to mark an earlier stage than does FDG-PET. In his study, all 56 AD patients, and 28 of 58 MCI patients, had robust PIB binding in cortical areas, reflecting a typical AD pattern. But reduced glucose metabolism accompanied this high PIB uptake in only 29 AD and in merely two MCI patients. Among the 91 healthy seniors in this study, 17 had elevated brain amyloid; their PIB patterns had lower intensity but otherwise resembled those of the PIB-positive MCI and AD participants. None of these 17 healthy seniors with high PIB had reduced glucose metabolism. Across all groups, higher PIB load correlated with worse cognitive test performance (Mini-Mental State Examination and CDR sum of boxes). Taken together, Hatashita’s data suggest that brain amyloid may track poorly with glucose hypometabolism, but nonetheless holds promise as a means for diagnosing preclinical AD, Hatashita said. In a post-meeting conversation with this reporter, he proposed a model of AD whereby amyloid deposition strikes early and would require another “hit” (e.g., mitochondrial abnormalities, free radical damage, calcium dysregulation) to produce neuronal dysfunction detectable by FDG-PET.
In an effort to bring some closure to what was becoming an increasingly complex story regarding PIB-PET and FDG-PET, Jagust asked during a Q&A if these two measures are correlated, anti-correlated, or have no relationship. “It depends on where in the brain you look, and when you look,” Dawn Matthews offered in reply. Matthews heads Abiant, Inc. in Deerfield, Illinois, a company that conducts imaging studies to help pharmaceutical companies assess their drugs. She and collaborators at New York University identified 50 participants of the Alzheimer’s Disease Neuroimaging Initiative (ADNI) who had both FDG and PIB-PET scans, preferably at least two PIB reads. In her talk, Matthews reported that her team saw progressively lower glucose metabolism levels in particular brain areas when comparing baseline scans of participant groups with increasing degrees of clinical decline. That is, normal controls had the highest metabolism levels; then came normal controls who converted to MCI; then MCI who stayed MCI; then MCI who converted to AD; and AD, whose metabolism was lowest. “Higher amyloid load and lower glucose metabolism seem to be correlated in regions known to be affected first in each modality, even in normal subjects,” she said. To explain, Matthews said via e-mail: “We did not find correlations between increased amyloid…and decreased glucose metabolism within medial frontal gyrus or prefrontal cortex, two of the primary regions in which amyloid has been found to accumulate. However, there was a correlation between amyloid in those regions and decreased glucose metabolism in the hippocampus (a brain structure involved in memory that is among the regions of earliest and most substantial atrophy in AD), as well as other regions where glucose metabolism declines in AD.”
Klunk parried Jagust’s question in the group Q&A with a question of his own. “Could increased basal metabolism to begin with accelerate amyloid deposition?” he asked, noting work by Washington University researchers that links higher synaptic activity to Aβ release into the interstitial fluid (see ARF related news story and Cirrito et al., 2005). Klunk then followed with this proposal: If people start at a high level of metabolism and can maintain that, perhaps it takes more of a second insult, e.g., Aβ, for them to progress from the MCI group into AD. Earlier in the day, Mormino offered a similar temporal argument for hypometabolism. “Maybe if your metabolism is slow and then you get hit by amyloid,” she said, you become more susceptible to decline. Then again, she said, “it could be that the amyloid induces the FDG-PET changes down the line.” Not to get lost in uncertainties, it is worth noting that this notion crystallized for the HAI conference participants: Amyloid deposition correlates with functional brain changes. See Part 2 of this series for more.—Esther Landhuis.
This is Part 1 of a four-part series. See Part 2, Part 3, and Part 4.
All attendees are invited to send additions and corrections to esther@alzforum.org.
Figuring out what high levels of brain amyloid mean for otherwise healthy older people was a prime focus of the Human Amyloid Imaging (HAI) conference, held 24 April in Seattle. Along with new research suggesting correlations between high amyloid load and declining glucose metabolism (see Part 1 of this series), several other studies strengthen the case that amyloid-laden brains are on the wane. These data help relate live measurements of Alzheimer disease (AD) pathology to subtle changes in brain function that may be present years before standard tests pick up cognitive decline. Understanding what happens during the critical time window between early pathology and possible dementia onset should help open the doors to future preventive therapies for AD, researchers at the conference agreed.
Like several other speakers, John Becker, Massachusetts General Hospital, Boston, tackled this basic question: Does amyloid deposition correlate with diminishing brain metabolism in cognitively normal older people? Becker measured both features with positron emission tomography (PET)—the former with the amyloid tracer Pittsburgh Compound-B (PIB), the latter as fluorodeoxyglucose (FDG) metabolism—but his FDG-PET assessments focused on brain areas that make up large-scale functional networks. Among 53 participants with a Clinical Dementia Rating (CDR) of 0, 11 had high PIB uptake. As a group these folks had lower glucose metabolism in portions of the default-mode network, including the posterior cingulate-precuneus, inferior parietal, lateral temporal, and ventromedial areas. The default-mode network is a system of brain areas that fire up during rest and tone down when the person focuses on a mental task. These same brain regions are among those hit first in AD (see ARF related news story and Buckner et al., 2005), and loss of default-mode activity in the resting state seems to mark early AD (see ARF related news story and Greicius et al., 2004).
Trey Hedden, a Massachusetts General Hospital colleague and collaborator on Becker’s work, used a different approach to address a similar issue. His study did not use FDG-PET but rather functional magnetic resonance imaging (fMRI) to probe the relationship between increased Aβ burden (measured by PIB-PET) and changes in the default-mode network. In Seattle, Hedden reported that healthy seniors with high PIB uptake had reduced functional coherence in the default network. This trend appeared in fMRI scans taken during rest and mental tasks, albeit more strongly at rest. Furthermore, higher amyloid load correlated with lower default-mode connectivity, even though the high-PIB and low-PIB groups appeared equal on memory tests and clinical assessments.
In another study presented at HAI, subtle cognitive differences that went undetected on standard neuropsychological tests did in fact show up in a more challenging memory test. Dorene Rentz, of Brigham and Women’s Hospital in Boston, tested whether amyloid load correlates with cognitive performance, and whether cognitive reserve influences this relationship in normal elderly. Often measured by years of education, or in this study by the AMNART IQ test, “cognitive reserve” seems to help protect against dementia in the face of brain Aβ accumulation (ARF related news story).
In Rentz’s study, which included 66 normal elderly and 17 AD patients, those with higher amyloid load in the precuneus area of the brain tended to fare worse in neuropsychological testing. When considering only the normal seniors, though, precuneus Aβ burden did not correlate with test performance unless a more difficult measure (Memory Capacity Test) was used. The MCT requires subjects to learn two 16-word lists, and it was the second list learning that differentiated the high- and low-PIB normal groups, Rentz reported. In her study, the relationship between precuneus amyloid load and cognitive test performance was weakened by cognitive reserve. “People with high cognitive reserve were able to maintain their performance, whereas those with low reserve were doing worse,” Rentz said. However, “at a certain point, cognitive reserve is no longer protective” and probably “loses its protective value after a certain level of amyloid is endured,” she noted. Overall, her data underscore the potentially confounding effects of cognitive reserve in neuropsychological performance, and suggest that more challenging tests are needed to pick up subtle cognitive deficits that escape standard measures.—Esther Landhuis.
This is Part 2 of a four-part series. See Part 1, Part 3, and Part 4.
All attendees are invited to send additions and corrections to esther@alzforum.org.
No Available Comments
The feast of amyloid- and PIB-centric data at this year’s Human Amyloid Imaging (HAI) conference, held 24 April in Seattle, came with morsels of alternative fare reminding attendees that Aβ and Pittsburgh Compound-B may not have the last word in Alzheimer disease research. This story describes a few such presentations.
Though PIB has shown great promise as an in-vivo amyloid tracer in brain imaging using positron emission tomography (PET), it is conceivable the compound does not pick up the full range of pathological Aβ in the brain. That idea came through in a talk by Chet Mathis, of the University of Pittsburgh in Pennsylvania, who co-invented PIB with colleague Bill Klunk. To gauge the threshold of Aβ plaque burden needed for a positive PIB-PET signal, Mathis’s team measured Aβ load in postmortem brain tissue from two people: the woman with severe AD from a recent paper by him and colleagues (Ikonomovic et al., 2008), and a 79-year-old man diagnosed with dementia with Lewy bodies (DLB). The amount of amyloid required to detect a PIB signal in the AD patient was 0.3-0.4 μM, and in the DLB patient it was even higher (i.e., more than 0.8 μM). Furthermore, the PIB binding seemed to be qualitatively different in the DLB subject, Mathis noted in a post-meeting e-mail to this reporter. Despite a negative PIB-PET scan a year and a half before this man’s death, the researchers found substantial numbers of neocortical plaques (mostly diffuse) and moderate concentrations of insoluble Aβ1-42 peptide in his autopsy brain samples. This illustrates how PIB uptake could be misleading, by showing that PIB-PET scans can come out weakly positive despite extensive Aβ deposition. Furthermore, the mismatch between PIB signal and Aβ plaque numbers suggests that 11C-labeled PIB may be exquisitely selective for unique confirmations of human Aβ, Mathis said.
That notion seems to jibe with recently published work in non-human primates, and with HAI data presented by Jitka Sojkova, a postdoctoral fellow in Susan Resnick’s group at the National Institute on Aging in Bethesda, Maryland. In the former investigation (Rosen et al., 2009 and commentary), cortical extracts from aged squirrel monkeys, rhesus monkeys, and chimpanzees had markedly lower PIB binding compared with human AD patients, even though their brain extracts had AD-like levels of insoluble Aβ. In Seattle, Sojkova described her efforts to relate in-vivo brain amyloid imaging with neuropathological Aβ assessments in cognitively normal seniors. She and colleagues studied six people who were enrolled in the Neuroimaging Substudy of the Baltimore Longitudinal Study of Aging. In the people who had very low or very high amyloid load, PIB-PET imaging correlated well with neuropathology studies done on average 1.5 years after their last PIB scan. However, for participants with intermediate PIB-PET uptake, “it's more complex,” Sojkova said. “It depends on the region we are looking at, the time to autopsy, and many other issues.” These findings earned Sojkova this year’s HAI Young Investigator Award. The award was available to conference attendees enrolled in or less than five years out of graduate school, medical school, or residency programs, and came with a U.S. $500 cash prize.
Even before Mathis and Sojkova had presented their data revealing limitations of PIB, Marsel Mesulam, of Northwestern University Feinberg School of Medicine in Chicago, Illinois, had done his part to shake up the amyloid-centric focus of the day’s discussion. His keynote address began with a tongue-in-cheek remark, noting that since his first AD-related publication in 1976, “I’ve managed to keep my lab running without writing a single paper on amyloid and without proposing any grant related to amyloid.” He said that though the genetics of AD favors an amyloid-based pathogenesis, neuropathology argues that neurofibrillary tangles (NFT) are the prime villain as they have a much higher density in memory-related parts of brain (e.g., nucleus basalis, amygdala, hippocampus). In addition, tangles kill the neurons within which they form, and this cell death tracks with cognitive decline, he said. These features make neurofibrillary tangles “one of the most reliable biomarkers of the age-MCI-AD spectrum,” said Mesulam. “The same cannot be said about amyloid.”
In particular, Mesulam noted that the relationship of plaques to neuropsychological state is anything but clear-cut. “In some patients, plaques are latecomers, so they can’t be blamed for the emergence of memory loss,” he said. Other patients, however, rack up loads of brain amyloid yet stay sharp. This is what Mesulam found when he analyzed the brain of an 80-year-old woman in the university’s SuperAging study (ARF related news story). In a postmortem brain section from this “superager,” i.e., a person 80 or older who performs like a 50-year-old on neuropsychological measures, “I was able to find just a single tangle (thioflavin S staining in entorhinal cortex) but thousands of plaques,” he said. “The only thing I can say is that maybe the toxicity of amyloid is overdone.” Mesulam did note that his arguments against amyloid were based on studies of fibrillar Aβ, though the real culprit may be circulating Aβ oligomers. However, “no one has shown yet that these circulating oligomers increase with age,” he said. “None of the existing imaging agents today is in a position to image these circulating oligomers.”
Notwithstanding such caveats, the field does have in hand 18F-labeled compounds that perform reasonably well as fibrillar amyloid tracers. These should eventually find their way into widespread clinical use because of their ease of production and longer stability relative to 11C-labeled PIB. Florpiramine (aka 18F-AV-45), the only 18F compound with data at HAI, is being developed by Avid Radiopharmaceuticals, Inc. in Philadelphia, Pennsylvania. Thus far, it has not been compared head-to-head with [11C] PIB, Mathis wrote in a post-meeting e-mail to ARF. “But from the data presented to date,” he noted, “it appears that [18F] florpiramine has about twice the non-specific signal of [11C] PIB in white matter and about 80 percent of the specific signal that PIB has in high plaque containing areas of the cortex.” Mathis added that based on available binding data, it appears that florpiramine and PIB share the same binding site(s) on fibrillar Aβ40 and 42. In a recent review (Klunk and Mathis, 2008), Klunk and Mathis acknowledged that PIB will not be a mass product, and suggested comparing the various 18F compounds each to PIB as a way of determining how the newer agents stack up against each other. (See also Q&A with Mathis below.)
A Phase 2 trial of [18F] florpiramine was completed in November 2008. As described on a poster at HAI, three trials at 20+ centers, totaling 93 healthy controls, 63 people with a clinical diagnosis of AD, and 60 with MCI, showed that 75 to 80 percent of AD subjects had high florpiramine binding on visual read, as did 40 to 50 percent of MCI cases and 15 to 30 percent of healthy controls. Ongoing trials are testing how this compound fares as a biomarker to detect amyloid changes induced by a number of experimental AD drugs, e.g., Pfizer’s 04494700, an oral compound formulated to prevent amyloid-β from binding to the RAGE receptor (receptor for advanced glycation endproducts), and Eli Lilly’s γ-secretase inhibitor, LY450139, aka semagacestat. Florpiramine is also being used in a study that will compare regional amyloid burden in Parkinson disease (PD) patients and normal seniors; in a Phase 3 study aiming to correlate [18F] florpiramine PET imaging and brain amyloid pathology; and in an observational study to determine if [18F] florpiramine PET scans can predict 18-month cognitive decline in healthy elderly and people with MCI or AD.
At HAI, Dan Skovronsky of Avid reported that APOE genotype influences amyloid burden, as measured by PET imaging with [18F] florpiramine, in MCI and AD patients as well as in cognitively healthy elderly. These results are consistent with preliminary data presented at HAI by John Morris. They also confirm a recent study of 28 non-demented seniors showing that fibrillar Aβ load as determined by PIB-PET imaging tracks well with AD genetic risk based on APOE genotype (ARF related news story and Reiman et al., 2009).—Esther Landhuis.
This is Part 3 of a four-part series. See Part 1, Part 2, and Part 4.
All attendees are invited to send additions and corrections to esther@alzforum.org.
Q&A with Chet Mathis. Questions by Esther Landhuis.
Q: Has it been determined that an 18F compound will be used in ADNI 2? If so, which one is the leading prospect?
A: Yes, an 18F-labeled amyloid tracer will be used in ADNI 2. No 11C-PIB studies are planned for ADNI 2 at this time. Avid's 18F-AV-45 is presently the lead candidate for ADNI 2. While Avid has been willing to give access to their tracer for ADNI 2 studies, GE Healthcare and Bayer/Schering decided not to give access to their tracers. AstraZeneca indicated that they would make their 18F-labeled tracer available to ADNI 2, but few people have seen AZ's data. They will present at ICAD this July. So Avid is the front-runner for ADNI 2 amyloid imaging studies at this time.
Q: Have any side-by-side comparisons been done using 18F-AV-45 and 11C PIB?
A: No, not to date. ACRIN (American College of Radiology Imaging Network) has funded a small study (10 elderly controls and 10 AD) to compare AV-45 to PIB at the University of Pennsylvania. ACRIN also funded Pittsburgh University to compare PIB and the 18F-labeled GE compound AH-110690 in 10 elderly controls and 10 AD. An NIH/NIA GO grant will be submitted this month to compare AV-45 to PIB in the ADNI PIB cohort (19 elderly controls, 65 MCI, and 19 AD).
Q: Based on the data so far, how do 11C PIB and 18F-AV-45 stack up against one another in terms of 1) sensitivity/reliability (i.e., what they detect) and 2) ease of use (e.g., availability, cost, and what's involved on the part of patients and staff)?
A: There have been no head-to-head comparisons yet. But from the data presented to date, it appears to me that AV-45 has about twice the non-specific signal of PIB in white matter and about 80 percent of the specific signal that PIB has in high plaque-containing areas of the cortex. So it is somewhat inferior to PIB, but it may be good enough for the job at hand. We shall see.
As for ease of use, AV-45 has clear advantages over PIB. It likely will be manufactured at a number of central locations and shipped to all of the ADNI sites that conduct PET studies (about 50 sites). PIB is only manufactured on-site at 12 ADNI sites. So including AV-45 in ADNI 2 could quadruple the number of PET sites doing amyloid imaging. That's a clear advantage that any 18F-labeled tracer has over any 11C-labeled tracer.
Q: Do AV-45 and PIB have the same binding site?
A: With the very limited amount of binding data available, it appears that the two compounds share the same binding site(s) on aggregated (fibrillar) Aβ40 and 42.
Q: Do you have a diagram that shows the chemical structure of the tracers in contention?
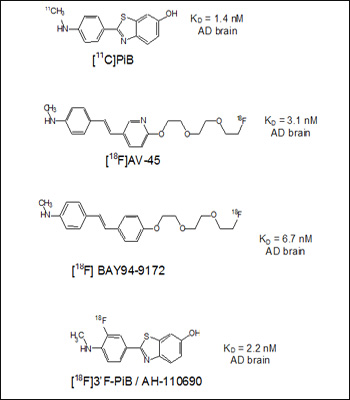
Four structures and reported affinities (Kd values) to AD brain homogenates. Image credit: Chet Mathis View larger image.
Note that PIB has the highest affinity and the Bayer compound the lowest (highest Kd). One atom, a nitrogen in the aromatic ring, separates AV-45 from the BAY94-9172 structure. Note also that the Bayer compound used to be called AV-1, until Avid licensed it to Bayer, so their similar structures are not a coincidence.
Q: Is there published data on AV-45?
A: At the present time, there is no peer-reviewed paper on AV-45 in the scientific literature. There are numerous abstracts and oral presentations on AV-45, but no published manuscripts. There are now more than 70 peer-reviewed literature papers on PIB. AV-45 has been in human use for about two years now.
Q: What happened to AV-1 (BAY 94-9172) and GE Healthcare’s 18F-labeled PIB derivative AH110690 (see ARF related news story)? Are these compounds still in the running or is most of the focus/energy going toward AV-45?
A: Both are still in the running of clinical trials. Bayer/Schering's compound was in Phase 2 studies some months ago. I haven't heard if they are completed. GE's compound finished Phase 2 trials and according to GET will enter Phase 3 trials in a few months. The two companies have decided to maintain "control" of their compounds by not making them available to ADNI 2. This is fairly standard for big pharmaceutical companies. Avid is a smaller, venture-funded company, hence in a different situation.
The earliest pathological underpinnings of Alzheimer disease often precede symptoms by untold numbers of years, presenting researchers with a two-edged sword. On the one hand, early intervention seems difficult when rogue proteins and pathways have ravaged the brain long before a person can tell a doctor something is amiss. On the other hand, optimists see in this long “latent” phase tremendous potential for new drugs to stop the disease before it gains a foothold. At this year’s Human Amyloid Imaging meeting, held 24 April in Seattle, the latter sentiment percolated through the day’s presentations and conversations. With continued advances in brain imaging techniques that measure atrophy, functional changes, and amyloid deposition in vivo, recognition is growing that the field by now indeed has worthy biomarkers in its arsenal, and may be poised to identify individuals at high risk for AD. This should help streamline clinical trials by providing quicker and more reliably measured endpoints, and refining selection of “normal” participants to those on the verge of cognitive decline. This part of our HAI series will summarize several biomarker presentations.
A question that has come to the fore in recent years is whether cognitively normal seniors with high levels of brain amyloid are in fact in greater danger of succumbing to AD than fellow seniors without amyloid. “Various biomarkers inform the issue…but they are not equal,” said Keith Johnson of Massachusetts General Hospital in Boston, one of the conference organizers. “Figuring out how these biomarkers relate to one another, and with cognitive change, will be important for disentangling the normal controls issue. Another thing that will be important is longitudinal data—how normals change over time,” Johnson said. This is critical to make the best use of biomarkers in future trials. “If you’re testing a drug in a big sample of people, you’re going to have a limited budget. You can’t run every marker on everybody. You have to understand how these biomarkers relate to each other to make an informed choice.”
In Seattle, John Morris of Washington University in St. Louis, Missouri, described a study designed to do just that. Preliminary analysis of its data suggests that the earliest biochemical hints of AD are not captured by amyloid imaging but by cerebrospinal fluid (CSF) biomarkers. This provocative conclusion emerged from a study of 241 cognitively normal seniors that used these two modalities to put into better perspective how age and ApoE genotype influence preclinical AD. By plotting amyloid burden (measured by the positron emission tomography [PET] tracer Pittsburgh Compound-B [PIB]) against age for subpopulations stratified by absence or presence of ApoE4 or ApoE2 alleles, Morris showed that ApoE genotype has a clear effect on PIB binding. This is consistent with a recent publication (Reiman et al., 2009 and ARF related news story). In Morris’s study, people who carried at least one E4 allele deposited amyloid with age more quickly than did those who lacked E4. For E2, the effect was protective: in people with an E2 allele PIB did not increase with age, Morris said. Similar trends appeared with CSF Aβ42, a biomarker that is believed to correlate inversely with PIB. Having an E4 allele accelerated the rate of decrease in CSF Aβ42 levels with age, while E2 carriers did not have this age-related decrease; if anything, their CSF Aβ42 levels edged upward as they got older.
When put through the same statistical gauntlet, CSF tau, unlike Aβ, did not seem to associate with ApoE4 genotype in normal elderly. To Morris, the study confirmed that the earliest pathological hints detected thus far in AD involve Aβ42 and not tau. “Tau abnormalities are important, of course, but occur later in the AD process, typically after dementia is fully expressed,” he noted in a post-meeting e-mail to ARF.
A case study from within this cohort of 241 provided further clues about the order of pathological events revealed, thanks to AD biomarkers. Two years before this man was diagnosed with very mild AD (Clinical Dementia Rating of 0.5) at age 90, his performance on episodic memory measures had begun a steady decline. However, the PIB-PET did not show elevated binding levels, and postmortem neuropathological analysis after his death at age 91 showed only small numbers of neurofibrillary tangles (Braak Stage II) and very few neuritic plaques or fibrillar plaques, the type detectable by PIB. The researchers did find numerous neocortical diffuse plaques, though, and CSF assays done at age 88.5 years showed markedly reduced levels of Aβ42, as well as minimal elevations of tau and phospho-tau.
Noting that firm conclusions cannot be drawn from a single case, Morris proposed nonetheless that this particular one “is consistent with a biomarker sequence for AD where the initial abnormality is represented by reduced levels of CSF Aβ42, corresponding to the deposition of diffuse amyloid plaques. Later, as the diffuse plaques become fibrillar, they also are detected by PIB and, as cognitive decline and dementia symptoms occur, by elevations of CSF tau and phospho-tau,” he explained.
In a paper published 11 May in the Archives of Neurology, work by a Washington University team that included Morris, David Holtzman, and first author Barbara Snider, among others, underscores the prognostic value of CSF markers. Studying 49 people with very mild AD, the researchers found that those with lower baseline CSF levels of Aβ42, higher tau or phospho-tau levels, or high tau:Aβ42 ratios, worsened more quickly in a follow-up assessment an average of 3.5 years later (Snider et al., 2009). Another recent paper by an independent group in Munich, Germany (Grimmer et al., 2009), confirmed that reduced CSF Aβ42 levels tracked with high PIB load by and large, but showed some regional differences.
At HAI, Cliff Jack of the Mayo Clinic in Rochester, Minnesota, offered his perspective on the sequence of AD pathological events as determined by serial PIB-PET and structural magnetic resonance imaging (MRI). Jack’s team assessed 21 healthy elderly, 32 people with amnestic mild cognitive impairment (aMCI), and eight AD patients in a longitudinal study involving clinical assessments, MRI, and PIB studies at two time points about a year apart. As previous longitudinal PIB-PET studies have shown, annual change in brain amyloid was small and about equal among all three clinical groups. In contrast, the rate of ventricular expansion, as determined by structural MRI, showed the expected trend (i.e., slowest for controls, intermediate for aMCI, and fastest for AD) and hence was able to distinguish the clinical groups. As for how these measures relate to cognition, PIB change did not correlate with performance in the CDR sum of boxes and only associated weakly with Mini-Mental State Examination (MMSE) scores. Here, too, ventricular expansion rates did correlate with cognitive decline as measured by these two tests. Jack proposed a model whereby amyloid accumulates at a slow, steady rate in late life—in contrast to neurodegeneration, which accelerates.
These data, published recently (Jack et al., 2009), suggest that amyloid imaging is useful for prediction while MRI is most useful for progression tracking during the clinical phase of AD, implying complementary roles for these two measures, Jack said. The same might be said for fluorodeoxyglucose (FDG)-PET and MRI. In a recent study using data from the Alzheimer’s Disease Neuroimaging Initiative (ADNI), FDG-PET appeared more sensitive at predicting cognitive decline in normal seniors, whereas MRI seemed to more aptly predict further decline for those who were already memory-impaired (Walhovd et al., 2008 and ARF related news story).
As for how changes in FDG-PET and brain amyloid load relate to each other during disease progression, Agneta Nordberg of the Karolinska Institute, Stockholm, Sweden, offered new insight (for more, see Part 1 of this series). At their five-year follow-up, members of an initial cohort of mild AD patients who had received PIB- and FDG-PET scans since 2002-2003 (see Klunk et al., 2004 and ARF related news story) showed very little change in PIB retention from levels at baseline and a two-year follow-up, Nordberg reported. This contrasts with FDG-PET results in these patients; their five-year cerebral glucose metabolism dropped considerably relative to the previous two time points. An ADNI data presentations meeting held in Seattle on 26 April, two days after HAI, featured more on the relative utility of various AD biomarker approaches. Stay tuned for the skinny on that.—Esther Landhuis.
This story concludes our conference series. See Part 1, Part 2, and Part 3.
All attendees are invited to send additions and corrections to esther@alzforum.org.
No Available Comments
Comments
No Available Comments
Make a Comment
To make a comment you must login or register.