Alzforum readers might be forgiven for thinking all microglia do is act prominently, if mysteriously, in Alzheimer's pathogenesis. Not so. A recent flurry of papers shows that microglia can match themselves specifically to GABA synapses. They can revive plasticity in the adult brain by “shaving” its extracellular matrix and making room for new spine growth. They snip away healthy synaptic spines in reaction to stress. Scientists are studying basic mechanisms by which these changeable cells may influence depression, PTSD. Do these mechanisms underlie some of the effects that have sparked renewed clinical trials interest in certain psychedelic drugs?
Series
Microglia in Health and Disease
New Ways to Target TREM2 Beg the Question: Up or Down?
Part 1 of 2
Microglia are the hot ticket in Alzheimer’s research these days, with a stream of studies showing that these cells can worsen or lessen pathology. In this balance, their receptor TREM2 acts as a pivot point, controlling a host of cell functions. Still, therapy-minded scientists remain unsure if it would be better to turn TREM2 up or down. So far, most research leans toward “up,” since pathogenic variants of TREM2 have reduced function. Now, work from Timothy Miller at Washington University in St. Louis casts a vote for “down.” In the July 6 Proceedings of the National Academy of Sciences, Miller and colleagues report that antisense oligonucleotides (ASOs) that squelch TREM2 expression halved plaque load in an amyloidosis mouse model. Surprisingly, belying other data, the drop in TREM2 seemed to activate microglial phagocytosis.
“There’s a brewing discussion about microglial states and whether they should be activated or inhibited, but the reality is that it is going to be disease-stage dependent and nuanced,” noted Kim Green at the University of California, Irvine. “We need better tools to address this, and these ASOs may help.”
It is also unclear what role the soluble sTREM2 fragment plays in microglial function, and what controls its shedding. In the June 25 Structure, researchers led by Alex Bullock at the University of Oxford, U.K., describe two antibody single-chain variable fragments (scFvs) that bind TREM2 and inhibit production of its soluble stub. The authors found that the antibodies dimerized before slowing TREM2 cleavage, and that they may act by stimulating cells to take up the receptor.
“The paper is a valuable contribution for the community, as it provides the first publication of the three-dimensional structure of TREM2-antibody complexes,” Kai Schlepckow at Ludwig-Maximilians University in Munich, Germany, wrote to Alzforum (full comment below). Like ASOs, scFvs may be valuable tools for TREM2 research, he added.
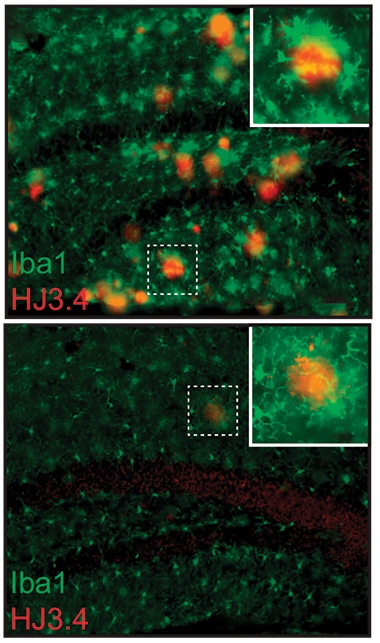
Less Active, Less Amyloid? Plaques (red) are numerous in the hippocampi of APP/PS1 mice (top). After TREM2 ASO treatment (bottom), plaques are sparse and microglia (green) more ramified, indicating a resting state. [Courtesy of Schoch et al., PNAS.]
TREM2 orchestrates microglial responses, boosting survival and phagocytosis to protect the damaged brain (Feb 2015 news; Jul 2016 news; Apr 2017 conference news). It also prods microglia to enter the disease-associated state seen around amyloid plaques in mice. When in this DAM state, the cells compact or perhaps even build plaques (May 2016 news; Sep 2017 news; Apr 2021 news). Several studies link TREM2 activation to fewer plaques but, confusingly, other studies report that TREM2 knockout lessens amyloidosis (Mar 2018 news; Dec 2014 conference news; Jay et al., 2017).
To disentangle this, first author Kathleen Schoch at WashU generated antisense oligonucleotides against mouse TREM2 and injected them into the ventricles of 10-month-old APP/PS1 mice. At this age, the mice have widespread plaques and highly activated microglia, as seen by Iba1 staining. A single jab with ASOs squelched TREM2 expression by 90 percent throughout the brain, and kept it low for longer than eight weeks.
One month after the injection, plaque load had dropped by half. Phosphorylated tau around plaques had fallen by a third. This p-tau may represent dystrophic neurites, the authors noted. Microglia around plaques looked less activated than in control mice, expressing less Iba1 and other pro-inflammatory markers, and having a more ramified shape (see image above).
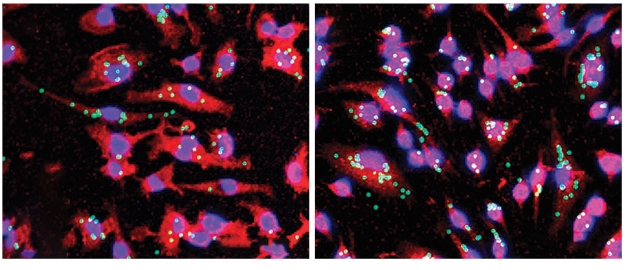
Better Eaters. Cultured microglia (red) expressing little TREM2 (right) consume more beads (green) than do controls (left). [Courtesy of Schoch et al., PNAS.]
How did ASOs produce this effect? At first, when the authors examined microglia one week after ASO injection, they saw a different picture. At this early point, microglia expressed more Iba1 and ApoE than in controls, suggesting the cells were activated. In keeping with this, isolated microglia from these mice gobbled up 50 percent more beads than those from controls, indicating enhanced phagocytic capability (see image above). The results suggest that knocking down TREM2 jolts microglia into a more phagocytic phenotype, spurring plaque cleanup. The subsequent drop in activation seen three weeks later may be a result of plaque clearance having quieted the microglia, Schoch wrote to Alzforum.
However, what the ASOs did depended on disease stage. ASOs injected at 4 months of age, before plaque deposition, or at 7 months, during plaque accumulation, had no effect on plaque pathology by 11 months. In other studies, too, manipulating TREM2 has different effects at distinct stages of disease (Jul 2018 conference news; Jan 2019 news; Apr 2020 conference news).
“Microglia are doing different things at different disease stages, and that needs to be considered when thinking about translation to humans, especially as pathology is occurring at different speeds and stages within the vastness of the human brain,” Green wrote to Alzforum (full comment below). Schoch agreed. “Our data would suggest that therapeutic targeting of TREM2 to alter microglial responses would be challenging,” she wrote.
The mice in this study had neither neurodegeneration nor cognitive decline at the age tested, so the study says nothing about whether inhibiting TREM2 to clear plaque would benefit these phenotypes. In addition, the mice do not accumulate tau, and some studies suggest TREM2 has opposite effects on amyloid and tau pathology (Oct 2017 news; Jul 2019 conference news; Jun 2020 news). Schoch plans to test TREM2 ASOs in mice with both pathologies.
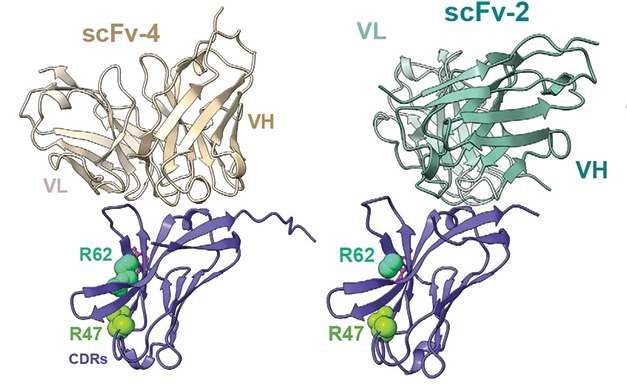
First Glance. Crystal structures of TREM2 (purple) bound to two single-chain variable fragments, scFv-2 (right) and scFv-4 (left). They bind TREM2’s immunoglobulin domain on the opposite side from the CDR regions that recognize ligands. [Courtesy of Szykowska et al., Structure.]
“While it remains unclear if targeting TREM2 is an appropriate therapeutic avenue, our study highlights the remarkable ability of microglia to transform the course of pathology in disease,” Schoch noted.
Most attempts to target TREM2 thus far have used antibodies, and two antibodies that activate the receptor are in clinical trials (May 2019 conference news; Mar 2020 news; Jun 2020 news).
Bullock and colleagues wanted to better characterize how TREM2 interacts with antibodies at a molecular level. To do this, first author Aleksandra Szykowska generated four scFvs against TREM2’s extracellular immunoglobulin-like domain, i.e., residues 19-131. Fragment 1 bound weakly and was set aside. Fragment 2 bound well enough to crystallize the complex and solve its structure (see image above). Alas, when added to cultured HEK293 cells expressing TREM2, scFv-2 did not affect sTREM2 shedding. Fragment 3 bound with higher affinity than scFv-2, and lowered sTREM2 by 22 percent. It dissociated quickly, however, so the crystal structure could not be solved.
Fragment 4 was the winner. It bound with high affinity and more specifically than did scFv-3, with less off-target binding. In culture, it lowered sTREM2 by 38 percent, meaning it was biologically active. The crystal structure of the complex showed that, like scFv-2, fragment 4 recognized an epitope on the other side of the immunoglobulin domain from the complementarity-determining regions (CDRs) that bind TREM2’s ligands. This implies that the antibody fragments would not interfere with ligand binding.
“Such binding sites would not be impacted by most AD risk variants, including R47H and R62H, suggesting a therapeutic potential of these scFvs to target TREM2 signaling,” noted Marco Colonna and Yun Chen at WashU.
Further probing revealed that scFv-3 and -4 affected TREM2 cleavage because they tended to form dimers. Purified monomeric preparations of fragments 3 and 4 had no effect, and fragment 2 did not dimerize, explaining why it was inert. “The need for scFv dimerization in order to inhibit TREM2 shedding is very much in line with what we have reported,” Schlepckow noted (Schlepckow et al, 2020). “It may be that both signaling and shedding of TREM2 involve receptor clustering,” Colonna and Chen agreed.
How do antibodies inhibit shedding? In cell culture, both scFv-3 and -4 caused cells to internalize TREM2, perhaps explaining how they prevent its getting clipped, the authors speculated. In contrast to these scFv fragments, the TREM2 antibodies in trials bind the stalk that connects the receptor’s immunoglobulin domain to its transmembrane portion. Thus, these antibodies may directly interfere with cleavage of sTREM2 (Aug 2017 news).
It is not completely clear if less TREM2 cleavage is good or bad in AD. Several studies have seen slower decline with high sTREM2, hinting cleavage is good (Jan 2016 news; Aug 2019 news). That said, is this because sTREM2 production reflects a high level of TREM2 signaling inside the cell, or because cleavage stops this signaling? If the latter, internalization of the receptor might accomplish the same thing. To boot, the soluble fragment on its own can also stimulate microglia, but again it is unclear if its effects are helpful or harmful (Feb 2017 news; Apr 2019 news).
Future studies may answer these questions. “We hope these additional tools will help elucidate the complex pathophysiology of TREM2 in AD,” Szykowska and colleagues wrote.
Lest a reader think microglia matter only in Alzheimer's, Part 2 of this series opens up a broader view on how these cells—through their ability to sculpt neuronal circuits—affect the brain in health and other diseases, particularly depression.—Madolyn Bowman Rogers
References
News Citations
- TREM2 Buoys Microglial Disaster Relief Efforts in AD and Stroke
- TREM2 Helps Phagocytes Gobble Up Aβ Coated in Antibodies
- New Evidence Confirms TREM2 Binds Aβ, Drives Protective Response
- Barrier Function: TREM2 Helps Microglia to Compact Amyloid Plaques
- ApoE and Trem2 Flip a Microglial Switch in Neurodegenerative Disease
- Microglia Build Plaques to Protect the Brain
- TREM2 Binds Aβ, Reprograms Microglia to Curb Plaques
- TREM2 Data Surprise at SfN Annual Meeting
- TREM2: Diehard Microglial Supporter, Consequences Be DAMed
- Without TREM2, Plaques Grow Fast in Mice, Have Less ApoE
- With TREM2, Timing Is Everything
- Changing With the Times: Disease Stage Alters TREM2 Effect on Tau
- TREM2, Microglia Dampen Dangerous Liaisons Between Aβ and Tau
- Boost or Block TREM2? Either Way, Therapy May Need Careful Timing
- Antibodies Against Microglial Receptors TREM2 and CD33 Head to Trials
- Paper Alert: Mouse TREM2 Antibody Boosts Microglial Plaque Clean-Up
- In Mice, Activating TREM2 Tempers Plaque Toxicity, not Load
- TREM2 Cleavage Site Pinpointed: A Gateway to New Therapies?
- TREM2 Goes Up in Spinal Fluid in Early Alzheimer’s
- In Alzheimer’s, More TREM2 Is Good for You
- Does Soluble TREM2 Rile Up Microglia?
- Cut Loose, Soluble TREM2 Beckons Microglia to Mop Up Plaques
- Not Just Alzheimer's: Microglia Sculpt the Brain in Health and Disease
Research Models Citations
Paper Citations
- Jay TR, Hirsch AM, Broihier ML, Miller CM, Neilson LE, Ransohoff RM, Lamb BT, Landreth GE. Disease Progression-Dependent Effects of TREM2 Deficiency in a Mouse Model of Alzheimer's Disease. J Neurosci. 2017 Jan 18;37(3):637-647. PubMed.
- Schlepckow K, Monroe KM, Kleinberger G, Cantuti-Castelvetri L, Parhizkar S, Xia D, Willem M, Werner G, Pettkus N, Brunner B, Sülzen A, Nuscher B, Hampel H, Xiang X, Feederle R, Tahirovic S, Park JI, Prorok R, Mahon C, Liang CC, Shi J, Kim DJ, Sabelström H, Huang F, Di Paolo G, Simons M, Lewcock JW, Haass C. Enhancing protective microglial activities with a dual function TREM2 antibody to the stalk region. EMBO Mol Med. 2020 Apr 7;12(4):e11227. Epub 2020 Mar 10 PubMed.
Further Reading
News
- Fall Flurry of Letters Kicks Up Dust Around TREM2
- TREM2 Mystery: Altered Microglia, No Effect on Plaques
- TREM2 Data Surprise at SfN Annual Meeting
- TREM2 Tidbits at AAIC: Genetics, Clinical Data
- United in Confusion: TREM2 Puzzles Researchers in Taos
- Unbiased Screen Fingers TREM2 Ligands That Promote Aβ Uptake
- Paper Alert: TREM2 Crucial for Microglial Activation
- Without TREM2, Microglia Run Out of Gas
- Model Morass? R47H Mutation Scuttles TREM2 Expression in Mice, Not People
- Down to Sex? Boy and Girl Microglia Respond Differently
- TREM2 Variants and CSF sTREM2 Levels Differ by Race
Not Just Alzheimer's: Microglia Sculpt the Brain in Health and Disease
Part 2 of 2
Judging by recent coverage on this site, Alzforum readers might be forgiven for thinking all microglia do is act prominently, if mysteriously, in Alzheimer's disease pathogenesis (see Part 1 of this series). Not so. Recent research has raised the profile of microglia overall, promoting them from garbage disposal crew to active sculptors of neuronal circuitry. Some basic biological functions coming to light now may become relevant to conditions as common as depression or post-traumatic stress disorder (PTSD).
Most recently, three new mouse studies shed light on how microglia prune synaptic connections during development, and how the same process can reawaken later in life—for good or ill. One study identified a subtype of microglia that express the GABAB receptor and trim inhibitory synapses. Such neuron-glia matching hints that their interactions may be as selective as neuron-to-neuron connections. Another paper describes how microglia can revive developmental plasticity: Stimulating the adult brain with the anesthetic ketamine or with flashing lights triggered microglia to chew up the extracellular matrix around interneurons. This created space that fresh synaptic spines then filled. Such a process might help the brain overcome negative experiences in PTSD or depression, and possibly even foreshadow microglial mechanisms underpinning the renewed interest in clinical trials of psychedelic drugs.
To be sure, stimulating microglia’s appetite can harm the brain, too. In the third study, systemic inflammation in youth somehow “stamped” microglia, priming them to over-prune synapses when the mice experienced stress later in life. This finding may help scientists elucidate how early life stress leaves the brain vulnerable to later neuropsychiatric disorders.
“These three studies add to the understanding of the essential functions of microglia in disease and neural plasticity, and they offer new information about specific mechanisms of action,” Douglas Fields at the National Institute of Child Health and Human Development in Bethesda, Maryland, wrote to Alzforum. Aviva Tolkovsky at the University of Cambridge, U.K., wondered whether the same fundamental mechanisms underpin each system. And would these be conserved in human brain? “These questions can only be addressed once the causal molecular players have been defined, several of which are newly proposed in these papers,” Tolkovsky wrote to Alzforum (full comment below).
Specialized Destruction: GABA Links Microglial Type to Inhibitory Synapses
Picky Eater. GABAB receptor-expressing microglia (green) contain within them synapses (red) from inhibitory neurons. [Courtesy of Favuzzi et al., Cell.]
Pioneering work from the late Ben Barres’ group at Stanford University first implicated astrocytes and microglia in synapse formation and maintenance, then found that synaptic pruning involved complement proteins of the innate immune system (Jan 2001 news; Dec 2007 news; Mar 2015 conference news). Microglia also devour synapses in response to environmental conditions, and in neurodegenerative disease (Nov 2010 news; Nov 2015 conference news; Oct 2019 news).

But how selective is this pruning? In the July 22 Cell, researchers led by Gord Fishell at Harvard Medical School revealed that microglia have a discerning palate for the synapses they eat. First author Emilia Favuzzi began by asking whether microglia could tell the difference between inhibitory and excitatory synapses. Inhibitory synapses respond to GABA, the brain’s premier inhibitory neurotransmitter (Shaye et al., 2021). Working with Fishell and Beth Stevens at Boston Children’s Hospital, Favuzzi fluorescently labeled inhibitory synapses in the somatosensory cortices of newborn mice and characterized the microglia that later engulfed them (see image above).
Surprisingly, perhaps, these microglia expressed the GABAB receptor, and they ignored excitatory synapses. They made up a quarter of all microglia in the region. Knocking out the GABAB receptor only in microglia produced a glut of inhibitory synapses by two weeks of age, but no change in excitatory synapses.
How would the GABAB receptor promote removal of inhibitory synapses? Partly by revving up the requisite machinery. Single-cell RNA-Seq showed that microglia lacking this receptor dampened expression of genes needed for synaptic pruning. This includes the complement protein C1q, which tags synapses for elimination. However, the scientists do not know how the system selects which inhibitory synapses to snip. Normal developmental pruning eliminates weaker, i.e., less active, synapses. “It’s counterintuitive, because GABA is the sign of a well-working synapse, yet it recruits microglia to prune synapses,” Fishell noted.
Curiously, though the GABAB receptor knockout mice had more inhibitory synapses than did wild-types at 2 weeks of age, by 2 months they had fewer. Mouse behavior reflected this, as seen by motion sequencing, a behavioral measure developed by co-author Sandeep Datta at Harvard. In this method, researchers film mice for an hour, tallying their different motions to develop fine-grained signatures of behavior. The 2-week-old knockouts were less active than wild-types, but by 2 months, they had become hyperactive. The data suggest a compensatory mechanism kicks in to offset the excess inhibition.
In future work, Favuzzi will delve into the mechanism behind inhibitory synapse pruning, and also search for microglial subtypes specialized to remove other types of synapses. Many neuropsychiatric disorders, including schizophrenia and autism, are marked by imbalances between excitatory and inhibitory synapses. If science could harness the brain’s immune system to restore this balance, it might help correct such disorders, Fishell suggested.
Gek-Ming Sia at the University of Texas, San Antonio, agreed this has potential. “This important work elegantly demonstrates a new idea. It opens up the intriguing possibility that modulation of microglia may be used to selectively tune specific synapses in a neural circuit to achieve a behavioral effect,” Sia wrote to Alzforum.
Siccing Microglia on Extracellular Matrix Unlocks Plasticity
Paring back synapses is a normal part of sculpting circuitry, but what about enabling new synapses to grow? Microglia apparently can do this, too. Previous research has shown that microglia need to be present for adult mice to learn new behaviors (Dec 2013 news). Microglia promote learning by trimming away extracellular matrix to allow new synapses to form (Jul 2020 news).

Vanishing Act. The perineuronal net (black) around interneurons in somatosensory cortex is thick under normal conditions (left), thinner after two ketamine treatments (middle), and gone after three (right). [Courtesy of Venturino et al., Cell Reports.]
In the July 6 Cell Reports, researchers led by Sandra Siegert at the Institute of Science and Technology in Klosterneuburg, Austria, focused on the role of the perineuronal net. The PNN is a sugary web that enmeshes parvalbumin-positive GABAergic interneurons; it constitutes one of the “tougher” parts of the extracellular matrix. The PNN locks synaptic circuitry into place (Pizzorusso et al., 2002; Frischknecht and Gundelfinger, 2012; Lensjø et al., 2017).
Enzymatic dissolution of the PNN is known to reactivate plasticity (Mar 2013 conference news), but Siegert wanted to experiment with the PNN in ways that are less destructive to the brain. She was intrigued by reports that low doses of the anesthetic ketamine, given to rats to induce schizophrenia-like brain changes, also thinned out the PNN (Matuszko et al., 2017; Kaushik et al., 2021).
To explore how this might work, first author Alessandro Venturino treated adult wild-type mice with high-dose ketamine. Three treatments completely abolished the PNN in the somatosensory and visual cortex (see image above). The strength of the effect was surprising, Siegert noted. “It’s rare in science that you see such a black-and-white phenotype,” she told Alzforum. As expected, removal of the PNN enabled new synapses to grow. When the scientists covered one of the animal’s eyes after PNN clearance, neurons connected to the open eye fired more, indicating synapse formation.
Importantly, the researchers traced the vanishing PNN to microglia. After ketamine exposure, microglia sidled up to interneurons and released matrix metalloprotease 9, an enzyme that degrades PNN. Chunks of the material appeared inside microglial lysosomes. By contrast, when microglia were depleted from mouse brain prior to ketamine exposure, the PNN stayed put.
The findings are evocative of the recent explosion of interest in ketamine as a potential treatment for conditions such as intractable depression, PTSD, and chronic pain (Carboni et al., 2021; Liriano et al., 2019; Cohen et al., 2018). Low-dose ketamine in the form of a nasal spray has been approved by the FDA to treat depression. Ketamine’s benefits are usually ascribed to its ability to block NMDA signaling, but the new data strengthen the idea that it may act by stimulating new synapse growth, as well (Krystal et al., 2017). That said, ketamine also has serious negative effects, including transient schizophrenia-like symptoms, sedation, and disordered thinking; its use must be carefully monitored.
How else could microglia be prodded to mow the PNN lawn? Ketamine is known to alter brain network activity, strengthening gamma frequency oscillations (Pinault, 2008; Ahnaou et al., 2017; Castro-Zaballa et al., 2018). To see if directly modulating gamma activity could produce the same effect, the researchers shined flickering lights into the eyes of mice. Lights flickering at 60 Hz, but not 40 Hz, slashed the amount of PNN in visual cortex by nearly half. Removing microglia before light exposure prevented this, demonstrating that these cells were responsible.
Microglia are known to monitor neuronal activity via P2Y12, a G-protein coupled purinergic receptor, or GCPR, on the microglial cell membrane. P2Y12 senses the ATP/ADP energy balance given off by neuronal cell bodies (Dec 2019 news). In this new study, blocking P2Y12 kept the PNN in place after 60 Hz treatment, again identifying neuronal activity as the key mediator of this microglial response. It appears that changes in the neuron’s energy level can ring the alarm bell for microglia, triggering them to “shave” the PNN and permit new synapse growth.
Curiously, 40 Hz light, which did not affect PNN in this study, has been reported to induce microglia to gobble up amyloid plaques and is currently in trials for Alzheimer’s disease (Dec 2016 news; May 2019 news; Apr 2021 conference news). The data suggest microglia and neuronal circuits interact in sophisticated ways, Siegert said, whereby different frequencies might elicit different reactions.
Fields agreed that microglia are sensitive to the frequency of brainwave oscillations and will intervene to restore normal neural activity patterns. “Therapeutic applications that exploit the involvement of microglia in regulating excitability of neural circuits may be possible, but much more research is needed,” he wrote to Alzforum. Siegert speculated that, like ketamine, PNN removal by light treatment eventually could be harnessed to treat neuropsychiatric conditions such as PTSD or depression. Both safety and efficacy of either ketamine or light treatment require further study.
David Attwell, Pablo Izquierdo, and Hiroko Shiina at University College, London, noted that the P2Y12 receptor facilitates synapse elimination, too. Removing the PNN may aid microglia in this endeavor, because stripping away the sugary “beard” enables microglial processes to contact neuronal membrane more easily (Sipe et al., 2016; Imbert et al., 2021). “Removing PNNs isn’t a magical solution to devastating neurological disorders … there may be a fine balance between retaining PNNs to protect neurons from toxic substances, and removing them to create space for synaptic growth,” they cautioned (full comment below).
Microglia were recently reported to chew up PNN in Alzheimer’s disease, both in a mouse model and in human brain tissue (Crapser et al., 2020).
Ádám Dénes, Institute of Experimental Medicine, Budapest, Hungary, pointed out that P2Y12 mediates different microglial actions depending on whether the receptor contacts the neuronal cell body or its dendrites (Cserép et al., 2020). “We assume that microglia-PNN interactions may also be, to some extent, different in different cellular compartments,” Dénes wrote to Alzforum. “Untangling the molecular mechanisms of compartment-specific microglia-neuron interactions may be vital to understand how exactly microglia-mediated effects operate under physiological conditions and in brain diseases.”
Microglia, the Culprit in the Two-Hit Hypothesis
Not to get carried away by microglia’s benefits, these cells can clearly wreak havoc in the brain. Some scientists believe microglia mediate the “two-hit hypothesis” of neuropsychiatric disorders, whereby inflammation early in life leaves the brain prone to depression or schizophrenia during stressful experiences years later (Maynard et al., 2001; Giovanoli et al., 2016; Mottahedin et al., 2017).
In the August 18 Neuron, researchers led by Zhi Zhang at the University of Science and Technology in Hefei, China, elaborated on this idea. Joint first authors Peng Cao, Changmao Chen, and An Liu injected lipopolysaccharide into 2-week-old, wild-type mice to trigger inflammation. At 6 weeks of age, the scientists stressed these mice in various ways, such as by restraining them, subjecting them to loud noise, or to “social defeat.” Under these pressures, LPS-injected male, though not female, mice lost their taste for sugar and gave up sooner when hung by the tail or forced to swim. These behaviors are believed to be a mouse approximation for human depression. The authors used male mice for all subsequent experiments.
After LPS, microglia in the anterior cingulate cortex became activated, as seen by stronger Iba1 staining and a more amoeboid shape. The more activated the microglia were, the more pronounced were the mouse’s later depressive-like behaviors. Depleting or inhibiting microglia before applying stress prevented these behaviors.

Lost Under Pressure. Mice subjected to LPS-induced inflammation in early life (right) lost dendritic spines after adolescent stress, while control mice (left) didn’t. [Courtesy of Cao et al., Neuron.]
As in the other studies, microglia exerted their effect by influencing synapses. Live two-photon imaging revealed microglia in the anterior cingulate cortices of LPS-treated mice swallowing glutamatergic synapses after restraint stress. The “inflamed” mice ended up with about a third fewer dendritic spines than saline-treated controls (see image at right). As might be expected, their glutamatergic neurons fired fewer action potentials. Inhibiting microglia, or activating neurons, prevented the depressive-like phenotype after stress.
LPS is known to activate toll-like receptor 4 on microglia, prompting upregulation of the fractalkine receptor CX3CR1, as well as several proinflammatory cytokines (Gan et al., 2013; Papageorgiou et al., 2016). In this study, microglia in LPS-treated mice doubled CX3CR1 expression relative to controls. This receptor is involved in synapse pruning (Paolicelli et al., 2011; Zhan et al., 2014). In keeping with this, knocking down CX3CR1 before LPS injection prevented subsequent spine loss and depressive-like behavior, while overexpressing CX3CR1 exacerbated it.
The data fit a model in which early inflammation kicks off a lasting change in microglia, causing them to express more CX3CR1. During later stress, neurons release fractalkine, which binds CX3CR1 and triggers microglia to devour spines. In this way, early life inflammation can lead to chronic maladaptation of the brain, the authors suggest.
If so, then could modulating fractalkine or its receptor become a therapeutic strategy? Fields cautioned that this system has many critical functions, so more research would be required to figure out how to tweak it safely.
Understanding what role overly active microglia and loss of spines might play in depression is a growing field (for current review, see Enomoto and Kato, 2021). Earlier this year, scientists found that the psychedelic mushroom component psilocybin helps lift depression by strengthening excitatory synapses in the hippocampus (Hesselgrave et al., 2021). Since then, Alex Kwan and colleagues at Yale University School of Medicine, New Haven, Connecticut, found that tracking the growth of new dendritic spines with two-photon microscopy enabled them to document a lasting 10 percent uptick in spine density in the frontal cortex after a single dose of psilocybin (Shao et al., 2021). Psilocybin and other psychedelics such as ketamine are being investigated for the treatment of PTSD (Krediet et al., 2020). Together, the data hint that such compounds might exert their effects by tweaking microglia’s interaction with synapses.
Kwan noted that for both ketamine and psilocybin, the issue of the best dosage to use remains underexplored. Some studies suggest that a subanesthetic dose may be most effective for boosting neural plasticity, but more work is needed. The mechanisms behind plasticity also need further elaboration. “Why there is a convergent effect for ketamine and psychedelics is still a complete mystery. Microglia could be part of the mechanism, but more work needs to be done,” Kwan wrote to Alzforum.
Siegert noted that because of the proliferation of RNA-Seq data, scientists now have more tools than ever to dissect the molecular mechanisms by which microglia affect spines and synapses and, consequently, neuronal health and behavior. “These are exciting times for the research community,” she told Alzforum.—Madolyn Bowman Rogers
References
News Citations
- New Ways to Target TREM2 Beg the Question: Up or Down?
- Glia May Regulate Synaptic Formation and Transmission
- Paper Alert: Does the Complement Devour Synapses?
- Microglia Rely on Mixed Messages to Select Synapses for Destruction
- No Rest for Microglia: These Immune Cells Manage Healthy Synapses
- Microglia Control Synapse Number in Multiple Disease States
- Human Microglia Eat Synapses, More So in Alzheimer’s
- Beyond Neighborhood Watch—Microglia Nurture Synapses
- With IL-33, Neurons Tempt Microglia to Nibble At Synapses
- In Pursuit of Toxic Tau
- To Monitor Neurons, Microglia Talk With the Boss, aka the Soma
- Flashy Treatment Synchronizes Neurons, Lowers Aβ in Mice
- Gamma Waves Synchronized by Light: Good for Synapses, Memory?
- Does Synchronizing Brain Waves Bring Harmony?
Paper Citations
- Shaye H, Stauch B, Gati C, Cherezov V. Molecular mechanisms of metabotropic GABAB receptor function. Sci Adv. 2021 May;7(22) Print 2021 May PubMed.
- Pizzorusso T, Medini P, Berardi N, Chierzi S, Fawcett JW, Maffei L. Reactivation of ocular dominance plasticity in the adult visual cortex. Science. 2002 Nov 8;298(5596):1248-51. PubMed.
- Frischknecht R, Gundelfinger ED. The brain's extracellular matrix and its role in synaptic plasticity. Adv Exp Med Biol. 2012;970:153-71. PubMed.
- Lensjø KK, Lepperød ME, Dick G, Hafting T, Fyhn M. Removal of Perineuronal Nets Unlocks Juvenile Plasticity Through Network Mechanisms of Decreased Inhibition and Increased Gamma Activity. J Neurosci. 2017 Feb 1;37(5):1269-1283. Epub 2016 Dec 30 PubMed.
- Matuszko G, Curreli S, Kaushik R, Becker A, Dityatev A. Extracellular matrix alterations in the ketamine model of schizophrenia. Neuroscience. 2017 May 14;350:13-22. Epub 2017 Mar 18 PubMed.
- Kaushik R, Lipachev N, Matuszko G, Kochneva A, Dvoeglazova A, Becker A, Paveliev M, Dityatev A. Fine structure analysis of perineuronal nets in the ketamine model of schizophrenia. Eur J Neurosci. 2021 Jun;53(12):3988-4004. Epub 2020 Jun 25 PubMed.
- Carboni E, Carta AR, Carboni E, Novelli A. Repurposing Ketamine in Depression and Related Disorders: Can This Enigmatic Drug Achieve Success?. Front Neurosci. 2021;15:657714. Epub 2021 Apr 30 PubMed.
- Liriano F, Hatten C, Schwartz TL. Ketamine as treatment for post-traumatic stress disorder: a review. Drugs Context. 2019;8:212305. Epub 2019 Apr 8 PubMed.
- Cohen SP, Bhatia A, Buvanendran A, Schwenk ES, Wasan AD, Hurley RW, Viscusi ER, Narouze S, Davis FN, Ritchie EC, Lubenow TR, Hooten WM. Consensus Guidelines on the Use of Intravenous Ketamine Infusions for Chronic Pain From the American Society of Regional Anesthesia and Pain Medicine, the American Academy of Pain Medicine, and the American Society of Anesthesiologists. Reg Anesth Pain Med. 2018 Jul;43(5):521-546. PubMed.
- Krystal JH, Abdallah CG, Averill LA, Kelmendi B, Harpaz-Rotem I, Sanacora G, Southwick SM, Duman RS. Synaptic Loss and the Pathophysiology of PTSD: Implications for Ketamine as a Prototype Novel Therapeutic. Curr Psychiatry Rep. 2017 Aug 26;19(10):74. PubMed.
- Pinault D. N-methyl d-aspartate receptor antagonists ketamine and MK-801 induce wake-related aberrant gamma oscillations in the rat neocortex. Biol Psychiatry. 2008 Apr 15;63(8):730-5. Epub 2007 Nov 26 PubMed.
- Ahnaou A, Huysmans H, Biermans R, Manyakov NV, Drinkenburg WH. Ketamine: differential neurophysiological dynamics in functional networks in the rat brain. Transl Psychiatry. 2017 Sep 19;7(9):e1237. PubMed.
- Castro-Zaballa S, Cavelli ML, Gonzalez J, Nardi AE, Machado S, Scorza C, Torterolo P. EEG 40 Hz Coherence Decreases in REM Sleep and Ketamine Model of Psychosis. Front Psychiatry. 2018;9:766. Epub 2019 Jan 17 PubMed.
- Sipe GO, Lowery RL, Tremblay MÈ, Kelly EA, Lamantia CE, Majewska AK. Microglial P2Y12 is necessary for synaptic plasticity in mouse visual cortex. Nat Commun. 2016 Mar 7;7:10905. PubMed.
- Imbert PR, Saric A, Pedram K, Bertozzi CR, Grinstein S, Freeman SA. An Acquired and Endogenous Glycocalyx Forms a Bidirectional "Don't Eat" and "Don't Eat Me" Barrier to Phagocytosis. Curr Biol. 2021 Jan 11;31(1):77-89.e5. Epub 2020 Oct 22 PubMed.
- Crapser JD, Spangenberg EE, Barahona RA, Arreola MA, Hohsfield LA, Green KN. Microglia facilitate loss of perineuronal nets in the Alzheimer's disease brain. EBioMedicine. 2020 Aug;58:102919. Epub 2020 Jul 31 PubMed.
- Cserép C, Pósfai B, Lénárt N, Fekete R, László ZI, Lele Z, Orsolits B, Molnár G, Heindl S, Schwarcz AD, Ujvári K, Környei Z, Tóth K, Szabadits E, Sperlágh B, Baranyi M, Csiba L, Hortobágyi T, Maglóczky Z, Martinecz B, Szabó G, Erdélyi F, Szipőcs R, Tamkun MM, Gesierich B, Duering M, Katona I, Liesz A, Tamás G, Dénes Á. Microglia monitor and protect neuronal function through specialized somatic purinergic junctions. Science. 2020 Jan 31;367(6477):528-537. Epub 2019 Dec 12 PubMed.
- Maynard TM, Sikich L, Lieberman JA, LaMantia AS. Neural development, cell-cell signaling, and the "two-hit" hypothesis of schizophrenia. Schizophr Bull. 2001;27(3):457-76. PubMed.
- Giovanoli S, Engler H, Engler A, Richetto J, Feldon J, Riva MA, Schedlowski M, Meyer U. Preventive effects of minocycline in a neurodevelopmental two-hit model with relevance to schizophrenia. Transl Psychiatry. 2016 Apr 5;6:e772. PubMed.
- Mottahedin A, Ardalan M, Chumak T, Riebe I, Ek J, Mallard C. Effect of Neuroinflammation on Synaptic Organization and Function in the Developing Brain: Implications for Neurodevelopmental and Neurodegenerative Disorders. Front Cell Neurosci. 2017;11:190. Epub 2017 Jul 11 PubMed.
- Gan AM, Butoi ED, Manea A, Simion V, Stan D, Parvulescu MM, Calin M, Manduteanu I, Simionescu M. Inflammatory effects of resistin on human smooth muscle cells: up-regulation of fractalkine and its receptor, CX3CR1 expression by TLR4 and Gi-protein pathways. Cell Tissue Res. 2013 Jan;351(1):161-74. Epub 2012 Oct 20 PubMed.
- Papageorgiou IE, Lewen A, Galow LV, Cesetti T, Scheffel J, Regen T, Hanisch UK, Kann O. TLR4-activated microglia require IFN-γ to induce severe neuronal dysfunction and death in situ. Proc Natl Acad Sci U S A. 2016 Jan 5;113(1):212-7. Epub 2015 Dec 22 PubMed.
- Paolicelli RC, Bolasco G, Pagani F, Maggi L, Scianni M, Panzanelli P, Giustetto M, Ferreira TA, Guiducci E, Dumas L, Ragozzino D, Gross CT. Synaptic pruning by microglia is necessary for normal brain development. Science. 2011 Sep 9;333(6048):1456-8. PubMed.
- Zhan Y, Paolicelli RC, Sforazzini F, Weinhard L, Bolasco G, Pagani F, Vyssotski AL, Bifone A, Gozzi A, Ragozzino D, Gross CT. Deficient neuron-microglia signaling results in impaired functional brain connectivity and social behavior. Nat Neurosci. 2014 Mar;17(3):400-6. Epub 2014 Feb 2 PubMed.
- Enomoto S, Kato TA. [Stress Mediated Microglial Hyper-Activation and Psychiatric Diseases]. Brain Nerve. 2021 Jul;73(7):795-802. PubMed.
- Hesselgrave N, Troppoli TA, Wulff AB, Cole AB, Thompson SM. Harnessing psilocybin: antidepressant-like behavioral and synaptic actions of psilocybin are independent of 5-HT2R activation in mice. Proc Natl Acad Sci U S A. 2021 Apr 27;118(17) PubMed.
- Shao LX, Liao C, Gregg I, Davoudian PA, Savalia NK, Delagarza K, Kwan AC. Psilocybin induces rapid and persistent growth of dendritic spines in frontal cortex in vivo. Neuron. 2021 Jun 25; PubMed.
- Krediet E, Bostoen T, Breeksema J, van Schagen A, Passie T, Vermetten E. Reviewing the Potential of Psychedelics for the Treatment of PTSD. Int J Neuropsychopharmacol. 2020 Jun 24;23(6):385-400. PubMed.
Further Reading
News
- San Diego: MHC Class I and Complement—Holding Down Second Jobs in the Synapse
- Not Just “Glia”: Astrocytes Are Specialized Eating Machines, Not Oligodendrocyte Siblings
- Curbing Innate Immunity Boosts Synapses, Cognition
- Paper Alert: Microglia Mediate Synaptic Loss in Early Alzheimer’s Disease
- Microglia Prune Synapses in a Subtype of Frontotemporal Dementia
- The THIK and Thin of Microglial Surveillance
- ApoE Variants Modulate Astrocyte Appetite for Synapses
- Microglia Give Astrocytes License to Kill
- Sans Complement: Amyloid Grows, Synapses and Memory Stay
- Microglial Regulation and Function Scrutinized at Heidelberg Meeting
- Astrocytic IL-33 Signals Microglia to Engulf Synapses
- Clotting Protein from Blood Incites Microglia, and Synapses Die
- Do Tribes of Astrocytes Wage War on Synapses?
- Neuronal SRPX2 Spoils Microglial Appetite for Synapses
- Sans C9ORF72, Microglia Devour More Aβ Plaques. Synapses, Too.
- Glial Cells Refine Neural Circuits