On July 13, the day before the Alzheimer’s Association International Conference started in Los Angeles, 263 DIAN participants and their partners met in the same location with scientists, clinicians, research funders, and representatives of the association for a pre-meeting. They celebrated a shared commitment to use the scientific opportunities inherent in dominantly inherited Alzheimer’s disease toward a better understanding and clinical trials to prevent the disease. Four stories in this series showcase the resilience of DIAN participants, progress in AD genetics, a first look at emerging RNA-based therapies, and an update on DIAN clinical trials.
DIAD: Families from Argentina, Canada, Minnesota Rally a Global Community
“One day you will tell your story of how you’ve overcome what you are going through now, and it will become part of someone else’s survival guide.” Luciana, 29, DIAN participant.
“The disease might lie inside us, but so does the cure.” This line from Lindsay, a 23-year-old college graduate, captured the mood in the room when 263 people from 15 countries gathered for a day of news, commiseration, and a reinforced commitment to solving the problem of Alzheimer’s disease. On July 13, the day before the Alzheimer’s Association International Conference began, 139 sons, daughters, mothers, and fathers from families affected with dominantly inherited mutations in the APP or presenilin genes traveled to Los Angeles to meet with 124 researchers and staff in academia, pharma companies, the association, and the National Institute on Aging, including its director, Richard Hodes.
The meeting offered a mix of science and family news. Researchers presented science updates on the Dominantly Inherited Alzheimer’s Network observational study (DIAN-Obs), which started recruiting its first members in 2009. In particular, Alison Goate of the Icahn School of Medicine, Mount Sinai, New York, summarized what nearly 30 years of studying familial AD genes has taught scientists (see Part 2 of this series). Tim Miller of Washington University, St. Louis, explained a genetic therapy that may be coming down the pike for DIAN families, and panels of scientists from within and outside of DIAN research fielded audience questions (Part 3). WashU’s Randy Bateman reviewed how far the clinical trials platform DIAN-TU has come since its inception in 2012, and WashU’s Eric McDade polled family members about how best to handle questions of pregnancy in a primary prevention trial in people 18 and older that is ready to enroll (Part 4).
Lindsay as a baby, with her older brother and their father years before the latter's symptoms began.
For their part, the families displayed a deepening bond among those who have come to these annual meetings since the inaugural one in Washington, D.C , which drew 100 people (Aug 2015 conference news). Connected by their shared experience and DIAN, Millennials from Japan are becoming friends with others from the Netherlands, the U.S., and elsewhere. In LA, DIAN families initiated first-timers, one of whom said: “Finally, I am meeting people who understand how I feel.” They missed previous attendees, such as Dean, Ted (last names withheld to protect family privacy), and others, whose advancing disease and work commitments kept them kept them home this year.
In 2008, when the DIAN initiative was first funded, there was widespread doubt that the network would draw enough participants to achieve its goal of recruiting 240 participants into an observational cohort. Ten years in, the overall network stands at 562 between its observational cohort and its trials unit, DIAN-TU. DIAN researchers and staff increasingly use a registry as a channel not only for recruitment but also for communication with existing participants, issuing newsletters through it as well as calls to help try out new cognitive tests and answer survey questions.
The audience at this family conference marveled at how new people around the world keep coming forward and take initiative. In Los Angeles, four family members addressed the audience. Below are edited excerpts from three of them; Alzforum withheld last names and hometowns to avoid accidental identification of their families.
Dan, Minnesota
“Dear Alzheimer’s scientists, please see beyond the stoic mask we present to you. See our anger, sadness, fear. Our families have been torn apart.
“Mine is a Catholic family of six. We grew up in the ’60s and ’70s with sports, school, road trips, friends, and family. In 2005 my dad started showing symptoms. He was diagnosed and died in 2013. Around that time, though, my two older brothers showed symptoms, too. That was the start of a journey of understanding, acceptance, action. My brother learned about DIAN and went to the Columbia University site, but still I did not know how our disease was genetic. I just thought it ‘ran in families.’ When a cousin told me I had a 50:50 chance, I joined DIAN.
“I faced big decisions: Do I get tested? Do I find out the result? I decided right away. Now we know four of us carry the mutation, I don’t, one is unknown.
“I’ve been devastated by survivor’s guilt. I feel huge gratitude that I do not have to face the day-to-day degradation of my life, but have been wracked with the question of why I am not a carrier. I know the answer now: so that I can help my siblings. We had a family conference to decide what to do about it. I had plans, now I do this.
“We use humor to take the sting out of the chaos of 24/7 care. It is easy to get paralyzed by fear. Please have the courage to ask for help.”
Luciana, Argentina
“My mom was 50 when her symptoms appeared. Now she is 69, still at home, but has been bedridden for the last nine years. I am the youngest of five, and eight years younger than my closest sibling. I was 10 when her symptoms started. Growing up was hard, as I was watching my mom forget the things about life that I needed to learn. She used to be a magnificent math teacher, but couldn’t solve problems any more. She was losing the ability to cook, to write, to talk, and now even to walk and eat. Everything related to my life as a teenager—school, friends, boyfriend—I talked about with my father, who also took care of her. He passed away last December, leaving a huge hole in my heart. I don’t understand why life is so hard.
“Mom’s diagnosis was uncertain until a few years ago. In 2012, her sister died with similar symptoms and, together with my grandmother’s cause of death, made me distrust my mom’s initial diagnosis. My daughter was a baby and I began to fear the future. I did not want her to go through the same thing. I needed to find out what was going on. So I started to dig.
“It took me three years to piece together a family tree with 80 members across six generations. Nobody had done that before. As I am the youngest in my generation, some relatives had passed away long ago and it was difficult to find old people to talk to. Only a few provided data. I contacted distant relatives, introduced myself, and asked about neurological symptoms in them and their elders. I begged them to forgive that I was making them remember a painful past. I traveled to other provinces in Argentina, sat down with relatives there, and asked them about our relations. Then there was Susana, my mom’s cousin. I found her telephone number on the internet and called her. 'Are you Susana?' 'Yes.' 'Are you a daughter of Claudio?' 'Yes. Who are you?!' On our second phone call a year later, I realized Susana was symptomatic. Two years ago, she died. Her sister died at 49, of dementia. [Editor’s note: names changed to protect anonymity.]
“I visited hospitals, civil registries, and cemeteries to get dates, names, and causes of death. They were reasons like ‘psychological problem’ or ‘madness.’ In the pedigree, it looked as if the disease started near a great-grandfather, who was Lebanese. He arrived in Argentina at the beginning of the 20th century, where he raised four children, all of whom seem to have died young of dementia. I was able to get his death certificate and realized he died at age 51 in a psychiatric hospital. That made me think our family mutation came from Lebanon. I traveled there. I met family and got information about relatives, but nothing about the disease. So I believe he may have had the founder mutation.
“I took the pedigree to other doctors and showed them that my mom’s disease was not ‘just’ dementia. They ran a genetic test and found a mutation in PSEN1. The truth was devastating and a relief. If anyone in my family gets symptoms, we can know why and won’t have to start at the beginning.
“I felt responsible for others in my family who could be affected. But how would they react if I told them this sensitive information? I asked them first if they wanted to know our family’s genetic result, without saying that I already knew it. This way they could choose. Some did not want to know, others did but did not want to test themselves. I believe having information gives us power and opportunity; not having information leaves us unprotected and vulnerable.
“I hope my story encourages you. We are on the right path and an active part of the solution. All of you are now part of my extended family.”
Lindsay, Canada
“I am 23, and recently graduated from a university in Ontario. I’ll tell you how I chose to cope with my family’s Alzheimer’s disease. In eighth grade, I wanted to become a pharmacologist and find a cure for Alzheimer’s because I was watching it take my nana away
“Six of the nine siblings in my grandmother’s family had early onset AD. My dad showed symptoms at 44 and tested positive for a presenilin 1 mutation. I learned this the summer I left for university to study pharmacology. He is in the solanezumab arm of the DIAN-TU trial. He attended the family conference in London in 2017, but cannot travel anymore.
“At college, I joined my university’s AD society, raising money and educating peers about genetic forms of AD. Now I am on the board of Youngtimers, a nonprofit online resource some of us are building for young adults affected by this disease.
“My brother and I got life insurance, but are holding off on genetic testing. There is no DIAN-Obs site in Canada, and while there is a DIAN-TU site in Toronto, we are both too young for the DIAN-TU arms 1 and 2 that are finishing now. But we are in the right age bracket—above 18 and more than 15 years younger than the family age of onset—for the primary prevention trial that is starting up next year. I will be in that trial, 100 percent.
“When you join the primary prevention trial, you can’t get pregnant because it’s not known what the drug might do to a fetus. It’s extremely important to complete this trial and avoid drop-out, but by the time it’s done I will be at least 30. So I chose a fertility clinic as part of the internship requirements of my university programs. Working there helped me investigate freezing of my eggs, and also in vitro-fertilization, as a way to protect my ability to have children. I am in the middle of the process. I have finished the diagnostics, am taking supplements to improve the quality of my eggs, and this fall will take the necessary hormones and undergo the egg removal procedure.
“Choosing to freeze my eggs is one of the biggest decisions I made in life so far. It was a personal choice. I get anxious before the appointments, and it is expensive. [Editor’s note: in the U.S., egg freezing costs up to $20,000, plus an annual storage fee. Some employee benefits and health insurance policies may cover parts of it].
“Storing my eggs gives me a sense of control over this disease. And if I find out later that I carry the mutation, then I’ll have the option of preimplantation genetic diagnosis (Jul 2014 news series). Knowing how can protect my fertility while participating in PPT gives me comfort.
“When I graduated, it meant the world to me to have dad watch me walk across the stage, whether or not he understood what was happening. My goals have changed since eighth grade. I will be a pediatrician, not a pharmacologist. But I am still determined to help find a cure for AD. I will just do it through research participation instead.”
Lindsay and other family members, including Doug and Ione Whitney (from the Reiswig pedigree afflicted with a presenilin 2 mutation, see 2010 book review), were seen throughout the main AAIC conference that immediately followed the family meeting. For an update of genetics research in DIAN, see Part 2.—Framed and edited by Gabrielle Strobel
Each year, the DIAD family conference held in advance of the Alzheimer's Association International Conference features a science update, and this year the focus was on genetics. DIAN families know that the modern era of Alzheimer’s research started when blood given by their parents enabled the discovery of mutations in APP, presenilin 1 and 2 (Goate et al., 1991; Sherrington et al., 1995; Rogaev et al., 1995; Levy-Lahad et al., 1995). They may not know that Alzheimer’s genetics research today continues to draw on dominantly inherited AD. “The discoveries go back to the samples you donated in the 1980s, but they really do not end there,” Alison Goate told the families. Goate, who was at University College London when she discovered the first APP mutation, is now at Icahn School of Medicine, Mount Sinai, New York.
By the late 1990s, after scientists had fingered ApoE as a risk gene for late-onset AD (Corder et al., 1993), progress in AD genetics overall slowed to a crawl for a decade or so. Scientists knew that APP and presenilin mutations caused rare, dominantly inherited AD on the one hand, and that ApoE4 explained a small piece of the common, late-onset form of AD on the other hand. However, in between these two was a large unexplained gap they were unable to close with the techniques at the time. They were stuck. Then, with the advent of GWAS and new sequencing and analysis methods, AD genetics research revived—and once again, dominantly inherited mutations are part of the progress.
Nowadays, scientists use the term “genetic architecture” to convey that the genetic basis of Alzheimer’s disease is more like a complex assembly of many elements than the product of a single gene. The respective “power” of different gene mutations, or variants, falls onto a spectrum. On one end are dominantly inherited variants in a single gene that are powerful enough to cause AD in early adulthood; they are rare. On the other end are variants that raise a person’s Alzheimer’s risk by only a smidgen; they occur commonly among the different world populations. In between these extremes of severity and frequency, however, are known to be many variants in many additional genes that are more or less pathogenic and more or less frequent. This in-between group contains ApoE and many other genes, including Trem2, CD33, and Sorl1, for example (Campion et al., 2019). For every given person, it is their inherited unique combination of variants from this collection of genes that determine if and when they develop dementia.
This much-cited image from a paper by Teri Manolio and colleagues depicts how disease variants of genes fall onto a spectrum defined by strength (effect size) and frequency in the population. DIAD mutations would crowd the top left corner. Many of the weaker, and more common, gene variants that are still being discovered likely influence when Alzheimer’s dementia develops in a given APP and PSEN mutation carrier. [Manolio et al., Nature, 2010.]
How does this play out in reality? For example, the 501 families who are in DIAN all have a known pathogenic mutation in APP, PSEN1, or PSEN2, and almost all carriers get the disease. Even so, age of onset varies from one person to the next. This is especially true of PSEN1, which, at 392 families with 177 different mutations, is the most commonly mutated gene in DIAD. While many mutations cause symptoms by one’s 40s, the spread across PSEN1 mutations ranges from age 20 to 80, and even within a given family’s single mutation, onset can differ by a decade. Indeed, when scientists looked among non-DIAN families who had clusters of late-onset AD (LOAD), 3.4 percent of these people turned out to carry a “DIAD” mutation (e.g., Cruchaga et al. 2012). This means that other genes influence when a pathogenic APP or presenilin mutation breaks through in a person, Goate said.
One such gene is ApoE, insofar as APP/PSEN mutation carriers with the ApoE2 variant tend to get the family disease later than relatives with the ApoE4 variant. Another AD risk gene that preoccupies scientists is TREM2. It encodes a receptor protein sitting on microglia, the immune cells of the brain. What TREM2 risk variants do in DIAN families is unknown, but it is known that they speed up amyloid plaque formation in the brains of mice made to express DIAD mutations. Bits of the TREM2 protein are detectable in CSF of DIAN participants, and levels rise as people approach the symptomatic stage. “TREM2 probably influences progression of your family disease. The wide spread in age at onset that we see may be partly due to which TREM2 variant a person has,” Goate said.
Colored dots on this Manhattan plot represent spots in the genome where geneticists have identified Alzheimer’s risk. To date, they know of 41 such loci. [Courtesy of Shea Andrews & Brian Fulton-Howard.]
Besides ApoE and TREM2, many additional AD risk genes may play into age at onset. In toto, geneticists to date have identified 41 spots, or loci, along the human genome that harbor risk variants for late-onset AD (see image above). When they compute all of these variants into one number—called a polygenic risk score, or PRS—they can see that this score partially accounts for the age at onset of dominantly inherited AD. In other words, the PRS score does not determine whether a DIAN participant will develop AD. That is largely due to the powerful APP or presenilin mutations. Rather, the other AD genes combined affect at what age that happens (Cruchaga et al., 2018).
Using blood from both DIAN and API participants, geneticists are hot on the trail of protective genes. Addressing DIAN families in L.A., Ken Kosik of the University of California, Santa Barbara, emphasized that no gene mutation is 100 percent deterministic. Scientists know that besides ApoE2, certain variants of the genes TMEM106B, phospholipase C, Rab10, and the Icelandic APP mutation delay Alzheimer’s onset (May 2019 news; Tavana et al., 2019; April 2016 news). And they are sure that more are yet to be found.
The DIAN families include several “escapees” who remain cognitively healthy long after their family’s age at onset. One, Doug Whitney, is a regular at the family meeting; this year again, he was as well as ever despite his presenilin 2 “Volga German" mutation. The Colombian cohort includes at present seven carriers of the E280 mutation whose dementia did not start until old age, even though it usually begins in the 40s. Kosik and others are searching for the genes that held their disease at bay. One such gene was grist for the rumor mill at AAIC; its publication is currently embargoed but due out soon. About the others, all Kosik knows so far is that their protective variants do not lie in protein-coding regions. “They are more complicated to find, but we will,” Kosik said. Noncoding mutations tend to work by controlling the expression of other genes.
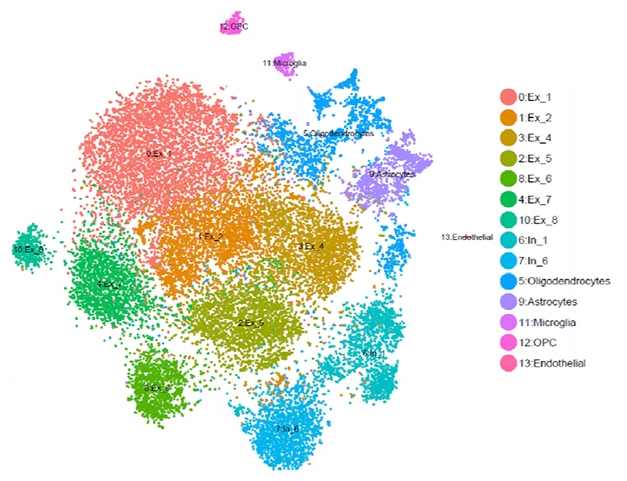
To understand gene expression in AD, DIAN is proving important in genomics research, as well. Genomics takes the study of genetics beyond a single gene. For example, scientists compare brain tissue from dominantly inherited to that of late-onset AD, and ask in which particular cell types a disease mutation does its damage. Is it neurons? Is it glial cells? At the DIAD family meeting, Goate cited studies by WashU’s Carlos Cruchaga, Oskar Harari, and others. They use pieces of postmortem brains donated by participants in DIAN and other AD studies, physically separate out the nuclei of the different cell types, and quantify which genes each individual cell had been expressing, and in what amounts, while the person was still alive. This is a step forward from mushing up bits of brain tissue and analyzing all the component cell types together, an older practice that obscured big differences between them.
Fewer Neurons in ADAD? An example of single-cell RNA analysis comparing brain tissues from a PS1 mutation carrier and noncarriers. Software named after the French pointillist painter Georges Seurat sorts gene-expression data into clusters. In this study, the carrier had fewer excitatory neurons (red, ochre, green) than noncarriers. Courtesy of Carlos Cruchaga and Oskar Hariri.
This type of genomics work is in its infancy, but so far, it appears that people with dominantly inherited AD have fewer excitatory neurons and more neuronal death at the same disease stage than people with late-onset AD (see image above; Del-Aguila et al., 2019; Li et al., 2018). If this trend holds up, it would confirm at the level of gene expression that dominantly inherited Alzheimer’s disease is a more aggressive form of AD. “These are examples of the work we do with the tissue you donate,” Goate told the families.
Mutations Keep On Coming
Incidentally, pathogenic APP and presenilin mutations are popping up anew in people at a rate scientists can measure. While current DIAN members have (or had) a parent with the disease, separate collections of sporadic, i.e. non-familial, early onset AD cases turn out to contain people who carry known pathogenic mutations in APP or PSEN1 or 2. In Los Angeles, Goate cited a study that included 10 such cases in whom that known mutation had newly arisen, i.e., the patient’s biological parents had not had AD (Lanoiselée et al., 2017). “These mutations look sporadic now, but in future generations they will look familial,” Goate said. Luciana, an at-risk DIAN participant living in Argentina, showed such an example at the family conference. A journalist herself, she traced her family’s mutation to her Lebanese great-grandfather. He likely suffered what looked like sporadic early onset dementia in his day, but passed on a presenilin mutation to at least 10 descendants to date (see Part 1 of this series).
In fact, a growing number of APP and presenilin mutations—some familial, some sporadic—have become known in Central and South American countries in recent years. Jorge Llibre-Guerra at WashU told Alzforum that people afflicted with the A431E PSEN1 mutation, first discovered in the Mexican state of Jalisco, may turn out to be as numerous as those carrying the E280A “Paisa” mutation in Colombia, for whom Francisco Lopera and his team at University of Antioquia in Medellin have been providing care, support, and now a first prevention trial. At 1,192 mutation carriers, the Colombian cohort is the largest known in the world today.
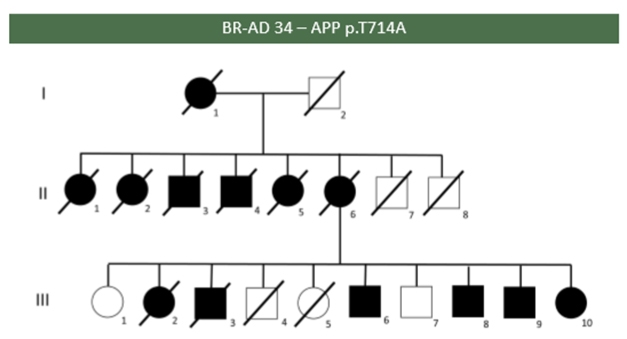
Affected (black) and unaffected members of a Brazilian family carrying the “Iranian” APP mutation T714A. Courtesy of Leonel Takada.
At the DIAD family meeting, Leonel Takada, University of São Paulo Medical School, showed a poster of the first Brazilian study assessing how frequently mutations can be found in autosomal-dominant AD at his clinic. Of the 17 probands from different families in Takada’s study, five had a mutation in PSEN1, of which four were previously described and one is new. Two probands from unrelated families had the same T714A APP mutation, known as the Iranian mutation. In one of the families, six of eight children of an affected mother had the disease, and the sixth sibling alone passed the mutation on to six of her 10 children (see image above).
Llibre-Guerra, Lopera, Takada, Ana Luisa Sosa-Ortiz of the National Institute of Neurology and Neurosurgery in Mexico City, and Ricardo Allegri of FLENI in Buenos Aires are working with the Alzheimer’s Association to obtain funding for a DIAN site to bring services, research, and eventually also prevention trials to families in this part of the world.
To learn about a genetic therapy for AD, see Part 3 of this series; for an update on ongoing and planned DIAN trials, see Part 4.—Gabrielle Strobel
Sherrington R, Rogaev EI, Liang Y, Rogaeva EA, Levesque G, Ikeda M, Chi H, Lin C, Li G, Holman K, Tsuda T, Mar L, Foncin JF, Bruni AC, Montesi MP, Sorbi S, Rainero I, Pinessi L, Nee L, Chumakov I, Pollen D, Brookes A, Sanseau P, Polinsky RJ, Wasco W, Da Silva HA, Haines JL, Perkicak-Vance MA, Tanzi RE, Roses AD, Fraser PE, Rommens JM, St George-Hyslop PH.
Cloning of a gene bearing missense mutations in early-onset familial Alzheimer's disease.
Nature. 1995 Jun 29;375(6534):754-60.
PubMed.
Lanoiselée HM, Nicolas G, Wallon D, Rovelet-Lecrux A, Lacour M, Rousseau S, Richard AC, Pasquier F, Rollin-Sillaire A, Martinaud O, Quillard-Muraine M, de la Sayette V, Boutoleau-Bretonniere C, Etcharry-Bouyx F, Chauviré V, Sarazin M, le Ber I, Epelbaum S, Jonveaux T, Rouaud O, Ceccaldi M, Félician O, Godefroy O, Formaglio M, Croisile B, Auriacombe S, Chamard L, Vincent JL, Sauvée M, Marelli-Tosi C, Gabelle A, Ozsancak C, Pariente J, Paquet C, Hannequin D, Campion D, collaborators of the CNR-MAJ project.
APP, PSEN1, and PSEN2 mutations in early-onset Alzheimer disease: A genetic screening study of familial and sporadic cases.
PLoS Med. 2017 Mar;14(3):e1002270. Epub 2017 Mar 28
PubMed.
ASOs: Wave of the Future in Alzheimer’s Therapeutics?
When participants in the Dominantly Inherited Alzheimer’s Network (DIAN) met in Los Angeles on July 13, they heard from their at-risk fellow travelers (see Part 1 of this series), learned how their tissue donations continue to advance AD genetics (Part 2), and got up to speed about how therapies based on that research are coming onto the scene. It’s not CRISPR quite yet, but RNA-based medicines, called ASOs, are in the clinic. Timothy Miller of Washington University, St. Louis, first told the audience of DIAN families, researchers, staff, and funders that other medical fields are increasingly looking to DIAN as a way to understand their own genetic forms of disease. Miller has developed gene-based therapies for ALS and frontotemporal dementia and, key to this audience, one that is in line to be tested by the DIAN trials unit (DIAN-TU) in the coming years.
Miller’s lab is devising drugs that consist of chemically modified, short nucleic acid sequences. Called antisense oligonucleotides, or ASOs, these drugs are designed to find a specific, matching RNA sequence inside of cells, and trigger their degradation (DeVos and Miller, 2013). This can turn down the expression of a troublesome gene, or turn up the expression of a needed gene. ASO therapies for the childhood diseases spinal muscular atrophy and Duchenne muscular dystrophy, as well as for a peripheral cardiomyopathy called transthyretin amyloidosis, are already FDA-approved (April 2019 news; May 2019 conference news). Investigational ASOs for Huntington’s disease and for ALS are in Phase 3 clinical trials.
In some cases, ASOs silence the mutant allele of a gene while allowing expression of the good copy. In other cases—and this is relevant to DIAN families—an ASO simply reduces the overall expression of a gene whose protein is part of the disease process. In this case that is MAPT, the gene for tau, the protein best known for the neurofibrillary tangles in AD. Miller’s group designed an ASO to bind to tau messenger RNA and intercept its translation, lowering the amount of tau protein in the brain. This approach appeared sensible because animal research suggested that lowering tau might benefit all cases of Alzheimer’s disease, familial and sporadic. By comparison, mutations in MAPT do not cause AD but rather frontotemporal dementia, and presenilins would require an ASO specific to each pathogenic mutation.
In mouse models of tauopathy, the tau ASO drug penetrated brain tissue and reversed neurofibrillary tangle pathology. The mice lived longer. “Our goal was to make these tauopathy mice stable, but the treatment did more. It made them better,” Miller told the audience (DeVos et al., 2017). In subsequent studies, the anti-tau ASOs reduced the amount of tau in the brains of non-human primates.
Miller partnered with Ionis Pharmaceuticals in Carlsbad, California, to develop the anti-tau ASO into an investigational drug. In 2018, Ionis partnered with Biogen to take its ASOs through clinical trials. Called Ionis-MAPTRx aka BIIB080, this drug is now in a Phase 1 clinical trial in 44 people with mild Alzheimer’s disease. The placebo-controlled study runs at 10 sites in Canada and Europe.
One site is at University College London, U.K. UCL researchers including Alison Goate, Nick Fox, and many others have cared for and studied people with familial dementias since the 1990s, and more recently built a unit to conduct innovative clinical trials for them. Called the Leonard Wolfson Experimental Neurology Center, it is expanding to be able to do more gene-based treatment studies, for example of ASOs. “The Wolfson center has changed our ability to do trials. It’s dedicated to conducting neurodegeneration trials early in the course of development. That is new. We have built a team and expertise in those studies,” its deputy director, Catherine Mummery, told the DIAN families in L.A.
Besides overseeing the tau ASO trial for Alzheimer’s disease at UCL, Mummery is already working with DIAN participants enrolled in DIAN-TU’s solanezumab and gantenerumab secondary prevention arms (Part 4 of this series). “Being part of DIAN-TU taught me what our patients go through when they are in DIAN. We learned that being patient-centered is very important. I try to apply that to other trials we do in sporadic AD,” Mummery said.
The tau ASO trial is about midway through. “The first person was dosed in our unit in November 2017. It was nerve-wracking for her, and for us, but it went well. We have done 16 doses of this drug at our unit, all without serious adverse events. We learn a lot about how to conduct these types of trial so that they are not so burdensome, and so that we give the medication in a comfortable way,” Mummery said.
The trial follows a multiple-ascending-dose format, where scientists give increasing doses to four cohorts of people, and wait between cohorts to see if the prior dose was safe. The trial’s first and second cohorts are complete, and the third cohort nearly so. Participants are being watched for potential on-target effects that would indicate that lowering tau is detrimental, for off-target effects that would indicate the ASO silences RNAs other than tau’s, and for side effects indicating the ASO activates the immune system in untoward ways. “These trials require a lot of safety monitoring. We are very cautious,” Mummery said.
Results are expected next year, both on safety and on whether the ASO lowered tau levels in the participants’ CSF. Determining whether lower CSF tau stops or slows the disease and its symptoms will require a Phase 2 trial, in which people are treated for longer than the three months the ASO is given in the current Phase 1 study, Frank Bennett of Ionis told the audience in L.A. A competitor of Ionis, Wave Life Sciences, is also starting to make ASOs against target RNAs in Alzheimer’s disease; this company dispatched a staff neurologist, Serena Hung, to the DIAD family conference. One idea for additional ASO therapies in AD is to silence expression of the APP gene.
Because ASOs do not cross the blood-brain barrier, both Ionis and Wave Life are currently delivering theirs intrathecally, in other words, by way of an injection into the spinal canal. The anti-tau ASO trial offers an open-label extension, at which point investigators see how much participants mind this invasive form of drug delivery. “Every person chose to continue to open-label; that tells us a lot about how tolerable this is,” Mummery said. Even so, she would like to transition to less-onerous delivery modes, for example using little pumps implanted under the skin.
The goal of delivering ASO therapies in a way that enables them to cross the blood-brain barrier is an active area of study. “It’s a challenging problem that we have not solved yet,” Bennett told the families. But even though intrathecal injection is cumbersome, Miller insisted that people tolerate it well in ALS, Huntington’s, and other ASO indications. “Our goal is to develop drugs that have a major effect and are therefore worth delivering in this way,” he said.
This ASO is but one of a handful of drugs the DIAN-TU drug selection committee are currently evaluating for a series of tau-based biomarker trials they intend to run beginning in 2020. The goal, said Randy Bateman of WashU, is to have sufficiently well-understood amyloid- and tau-based therapies in hand for combination trials.
And what about CRISPR? Two years ago at the DIAD family conference, a young woman stood up and asked when CRISPR therapies would repair her family’s mutation directly in their DNA. “CRISPR is a fantastic idea, but not quite ready for human trials in Alzheimer’s disease,” Miller said in L.A. Research on it is happening. For example, WashU’s Celeste Karch is using CRISPR to repair the mutations in neurons cultured from induced pluripotent stem cells (iPSCs), which she derived from DIAN participants’ fibroblasts. The DIAN genetics core has banked some 100 fibroblast lines spanning 34 APP and presenilin mutations as a resource for CRISPR studies, among others. These lines were made possible by skin biopsies donated by DIAN family members, and they are available to other labs for study.
But that is just a start. Scientists still need to work out how to ensure the CRISPR repair mechanism cuts DNA only at the site of the pathogenic mutation. They also need to figure out how to get a sufficient amount into the brain, and show that irreversibly editing the DNA in a person’s brain cells is safe.
Even so, CRISPR therapy is making progress on diseases that are considered lower-hanging fruit, in part because their targets are easier to reach, such as the blood or the eye. The first human success story came out this week, in a woman with sickle-cell anemia (see NPR story). For this therapy, the women’s blood cells were treated with CRISPR outside her body, and reinfused. On July 31, the first in vivo CRISPR therapy—in which the gene editing happens inside the patient—started enrolling for its first clinical trial. This investigational medicine is injected into the eyes of children, in hopes it will repair a mutation in the CEP290 gene in their retina that leads to blindness. For an update on DIAN clinical trials, see Part 4.—Gabrielle Strobel
As DIAN Wraps Up Anti-Aβ Drug Arms, it Sprouts Tau, Primary Prevention Arms
At the fifth annual DIAD family meeting held July 13 in Los Angeles, Randall Bateman of Washington University, St. Louis, stood before an audience of laypeople who had come from around the world to hear the latest about the grand project to which they have for years been giving hours upon hours taking tests, lying in scanners, giving their blood and cerebrospinal fluid—and a whole lot of good faith. As of 2019, the Dominantly Inherited Alzheimer’s Network has been continually enrolling and cultivating trust and active engagement of families for a decade. The first secondary prevention trial that grew out of DIAN is coming to a close this year. What’s next, the families wanted to know?
The family conference was but one DIAN event around the Alzheimer’s Association International conference, held July 14–18. There were closed meetings of the DIAN observational study’s (DIAN-Obs) steering committee, of its trials unit (DIAN-TU), plus a dozen scientific talks and posters on DIAN data scattered throughout the AAIC conference itself. Even so, addressing the people whose family devastation Bateman and WashU’s John Morris set out to solve was key to keep the movement energized and maintain both recruitment of new people and retention of current members, despite the great effort and time involved.
To do that, Bateman laid out what lies ahead in clinical trials, and WashU’s Eric McDade worked with the 20- and 30-year-olds in the room in search of solutions for a primary prevention trial.
The initiative’s observational cohort and trials unit are separately funded, but integrated, projects. People who are in DIAN-Obs can transition into DIAN-TU when a treatment trial whose inclusion criteria they meet becomes available. DIAN-Obs, currently 562 people strong, works to recruit some 25 new members annually to compensate for attrition into TU or to advancing disease. DIAN-Obs has upgraded its procedures and data storage to meet good clinical practice and Code of Federal Regulations/FDA guidelines on electronic records, so that its samples can be transferred seamlessly and its data merged with those of DIAN-TU. The TU is tapping Obs for external control data, effectively increasing its placebo pool, whereas Obs is using TU placebo data to increase its sample size for progression studies.
DIAN-TU currently operates at 27 sites in eight countries, whereas DIAN-Obs operates at 19 sites, with Barcelona starting this fall. For example, Canada currently has four DIAN-TU sites but had no DIAN-Obs sites until now; McGill University in Montreal will soon start enrolling into DIAN-Obs. This difference reflects a lack of local funding for more DIAN-Obs sites. It also followed a realization DIAN leaders made when they increased outreach into additional countries. This brought forward many more families and their local neurologists than were known to exist at the start in 2008. Many of the newly found families, it turned out, were not ready to join DIAN-Obs, but did want treatment trials. Sixteen future DIAN-TU sites in Argentina, Brazil, China, Colombia, Japan, and Mexico are pending; three sites in the Netherlands and Germany are still facing some delays.
Since DIAN-Obs began, its research has defined the stages of Alzheimer’s disease beginning 20 years prior to symptom onset, and shown the order—at least as detectable with currently available markers—to be amyloid, tau, metabolism, brain shrinkage, memory loss, and dementia. DIAN received 235 data and tissue requests, fulfilled 80 percent of them, and 150 papers have been published with DIAN data. Starting in 2012, DIAN-TU, along with its sister project, the Alzheimer’s Prevention Initiative’s Colombia initiative, began the first AD prevention trials using anti-amyloid drugs and the first trials specifically for people with dominantly inherited Alzheimer’s disease.
Both DIAN and API have built a registry-driven platform to support a continual series of investigational drug trials that adapt as new research findings come out, for example by changing the dose, adding new biomarkers, and developing statistics suitable to their particular cohorts.
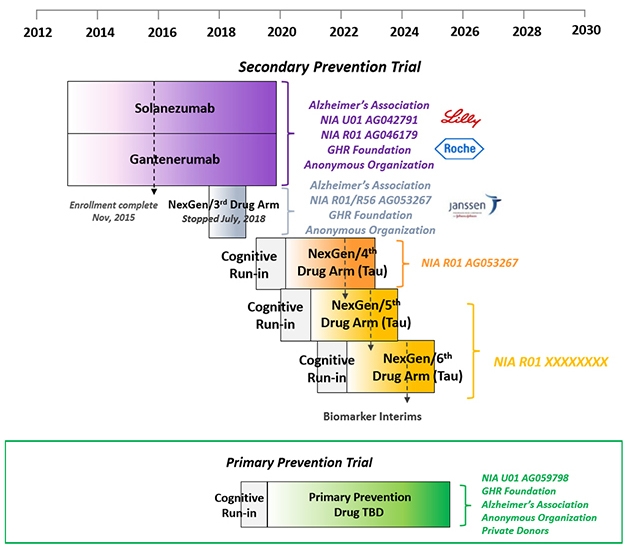
Having broadly defined stages of proteinopathy in autosomal-dominant AD, DIAN is set to conduct an ongoing series of primary and secondary prevention trials as participants, drugs, and funds become available. [Courtesy of DIAN-TU.]
The first two secondary prevention trials—of the anti-Aβ antibodies solanezumab and gantenerumab—will each see their last patient at the end of this year. Among themselves, DIAN participants are chatting about how to mark their last visit, and the nervous wait for the result after that. “Collecting and cleaning the data has started, and in 2020 we will work around the clock to analyze it,” Bateman told the audience in L.A., who included a fair number of participants in those two drug arms. “Once that is done, a group of people will be locked in a room and learn the results. Within 24 hours of that we will make an announcement to you and the world,” Bateman said.
DIAN-TU prevention trials platform. [Courtesy of DIAN-TU.]
(The third DIAN-TU drug arm, of the BACE inhibitor atabecestat, stopped in 2018 when its sponsor, Janssen, suspected liver toxicity. Indeed, at the AAIC conference in Los Angeles, David Henley of Janssen reported that further analyses of data from its other atabecestat trial in late-onset AD reinforced the decision to stop because atabecestat had worsened cognition slightly in people with very mild symptoms, as had other BACE inhibitors before it.)
What happens once the solanezumab and gantenerumab results are out, DIAN members want to know? If these two antibodies did not help, the trials stop and DIAN participants can consider the next trial, arm 4. If either worked, then its participants can join an open-label extension that will start next summer. One wrinkle here is that people will have to find out their mutation status at that point. Up to that point, they did not, because noncarriers were randomized to placebo. With open-label, however, noncarriers would expose themselves to side effects of a drug they do not need. Bateman recommended genetic counseling and testing after the placebo-controlled period, but only if a drug has proven effective, and said that DIAN will provide these services.
What about the next trials? Earlier this year, a cognitive run-in portion started for the fourth attempt at secondary prevention in people who have Alzheimer’s pathology but no symptoms. Ditto for a primary prevention trial in younger people who have no AD pathology yet. This run-in serves as a way for scientists and participants to get going now and strengthen the dataset with biomarker and progression data on each participant, even before a drug has been chosen. DIAN participants who are entering the fourth secondary prevention arm will have a tau PET scan as part of their run-in, as well.
Which drugs are right for the next trials? This pressing question is being debated behind closed doors. In L.A., DIAN scientists were not ready to say, except that the fourth trial will most likely test an anti-tau drug. Throughout the AD research field, calls for targeting tau therapeutically have been growing, though the first setbacks are starting, as well (Jul 2019 news). Recent PET data has shown that neurofibrillary tangles come up fast in familial AD around the time a person shows symptoms. This has given DIAN-TU researchers hope that tau PET scans will show them sooner than amyloid scans do whether a drug against tau has reached and reduced its target—and staves off symptoms. Curiously, tangles appear to spread differently in familial than sporadic AD; with a heavy presence in the back of the brain.
Tau PET scans suggest that neurofibrillary tangles form in the brain when a person with dominantly inherited Alzheimer’s becomes symptomatic, coming up faster and with a heavier burden in the back of the brain than in LOAD. [Courtesy of Benzinger et al., WashU.]
DIAN researchers believe that a big therapeutic effect is likely going to require hitting both Aβ and tau. Given that a handful of tau-targeting antibodies and other drugs, see below, have entered Phases 1 and 2 in LOAD, DIAN-TU is planning to evaluate three such drugs in the next five years in hopes of finding a winner for future combination trials. Drug arms 5 and 6, just like arm 4, will test a different tau drug in one- to two-year Phase 2 biomarker studies enrolling 70 people per trial. The best one will go forward into a Phase 3 trial that uses a cognitive endpoint and could support FDA approval, Bateman said. Funding for arm 4 is in hand, a grant proposal for arms 5 and 6 on track for submission this November. Bateman noted that unlike for arms 1 to 3, which were funded primarily by pharma, funding for arm 4 comes mostly from public and philanthropic sources, to afford the DIAN-TU drug evaluation committee full independence in their choice. Those funders include the Alzheimer’s Association, the GHR Foundation, and an anonymous organization.
The primary prevention trial, aka PPT, was widely assumed to evaluate a BACE inhibitor. Alas, CNP520, the Novartis drug being evaluated in a large Phase 3 secondary prevention program with API, stumbled over cognitive side effects earlier this month (Jul 2019 news). This latest downfall of another BACE inhibitor cast a pall over the future of the entire drug class. “This places great concern on anyone’s further use of BACE inhibitors,” Bateman said. One such inhibitor, elenbecestat, remains on track.
At AAIC, the prevailing outlook among most researchers about the prospects of BACE inhibitors was highly skeptical. That said, the cognitive decline seen at higher doses than would be used in primary prevention was small and did not worsen over time. Ease of use is an important consideration for a trial that runs for four to six years, and most antibodies require repeated infusion or injection, whereas BACE inhibitors come as pills. DIAN scientists are mulling whether a much lower dose of a BACE inhibitor is still an option, and until that is done, they are staying mum about which drug will go into the PPT.
This trial is a crown jewel at DIAN-TU, since it offers the purest test of the amyloid hypotheses attempted to date (Aug 2017 conference news). Last fall it received a large grant from the NIA and, subsequently, marching orders from the FDA. It aims to compare a drug to placebo in as many people as are necessary to enroll until 160 mutation carriers are on board. The precise number is unknown because genetic testing is not required to join. The blinded treatment phase will last for four years, with full site visits every two years, and short visits and frequent phone check-ins in between. Stopping amyloid accumulation is the primary goal. If the study drug does that, then all participants can go on open-label treatment for another two years, though again, at this point at-risk people would have to learn their mutation status.
This study faces unique design challenges. It invites all young adults at risk of a DIAD mutation, whether they know their genetic status or not, up until 15 years prior to their family’s age at onset. For example, if dad became symptomatic at 49, his children between 18 and 34 are eligible for the primary prevention trial (older children can join a DIAN-TU secondary prevention trial). The problem? Because exposing a fetus, or even sperm, to investigational medications is unacceptable, PPT participants must forgo pregnancy while they are on study medication, i.e., during their prime childbearing years—and half of them would do so while taking a placebo.
“We have been hearing from you that this is a serious issue,” McDade told the audience. To address it, McDade live-polled young adults at the family conference via an app to gauge what they would support.
In summary, four in five at-risk young people in the room indicated that they were willing to forgo pregnancy for the duration of the trial. Of the ones who were not, most said they would enroll if they could take a break after the four-year blinded phase. Conceivably, people could be in the trial, then get pregnant and go on open-label after the baby is born. Some people indicated they would prefer a “pregnancy break” after two years in the trial.
A quarter of the potential participants in the room had already considered family planning options such as freezing eggs for in vitro fertilization after the trial is over. Most indicated they would be interested in egg freezing, especially if the study was able to cover the expense. Lindsay, the 23-year-old daughter of a DIAN solanezumab trial participant, had told the audience earlier in the day how she had gone about egg freezing, and was mingling with other young people afterward (see Part 1 of this series). Most potential participants also endorsed meeting with a genetic and reproductive counselor as part of joining this trial.—Gabrielle Strobel








Comments
No Available Comments
Make a Comment
To make a comment you must login or register.