Skimping on Sleep Makes For More Aβ in the Brain
Quick Links
Mounting evidence suggests that the concentration of Aβ rises in people's cerebrospinal fluid if they don't sleep enough. However, scientists are unsure why that is. Do people make more of the peptide, or do they degrade less of it? A pilot study led by Brendan Lucey and Randall Bateman at the Washington University School of Medicine, St. Louis, now implies that enhanced production is to blame. As described in an Annals of Neurology paper published online December 8, volunteers who were kept awake overnight had a third more Aβ in their CSF by next day, compared with a relative dip seen in people who got a good night’s rest. While the number of participants was small, the study lends new clues to how sleep disruption could hasten a person along the path toward Alzheimer’s disease. “Chronic sleep deprivation or disruption could cause elevated concentrations of Aβ that, over time, increase risk of AD,” Lucey told Alzforum.
- In people who stay awake all night, CSF Aβ spikes by 30 percent.
- This is likely due to elevated synthesis, not clearance, of Aβ.
- Enhancing sleep with a drug didn’t lower Aβ levels.
Previous studies have linked disrupted sleep to future chances of developing AD (Yaffe et al., 2011; Tranah et al., 2011). Researchers have also found a rise in CSF Aβ levels in people who spent a single night without sleep, or in whom deep sleep was interrupted (Ooms et al., 2014; Ju et al., 2017). But it was unclear whether more synthesis or less degradation underlay the uptick.
To address the question, Lucey and colleagues used the stable isotope labeling kinetics (SILK) method Bateman developed to continuously monitor Aβ production and clearance from human cerebrospinal fluid (Jun 2006 news). Volunteers receive infusions of 13C-labeled leucine, which makes its way into newly made Aβ. The researchers use an intrathecal lumbar catheter to monitor the labeled peptide as it is synthesized and then cleared.
Lucey measured Aβ dynamics in this way in cognitive healthy volunteers aged 30–60 who had neither a sleep disorder nor a low Aβ42:Aβ40 ratio that would suggest they had Aβ deposits in their brains. Each person spent two nights in the lab. The first was spent acclimating to the new sleep environment, the second was the time of the experiment.
After the first night, each person was hooked up to a lumbar catheter at 7 a.m. The researchers monitored total Aβ levels by mass spectrometry until bedtime to estimate each person’s daily average. In general, Aβ concentrations started low in the morning and rose throughout the day, peaking at bedtime.
At 9 p.m., participants were infused with heavy leucine for the next nine hours. There were eight volunteers in total, and they all completed two or more sleep conditions, totaling 20 trials in all. At one time or another, seven people slept normally, six got a shot of sodium oxybate to enhance their deep, slow-wave sleep, and seven stayed awake by reading books, watching movies, or chatting with staff at the lab. Lucey monitored CSF C13 and total Aβ levels every two hours until 7 the next evening.
As expected, in all participants, the ratio of C13:C12 Aβ rose through the night and fell after the infusion was stopped. In people who slept, with or without sodium oxybate, total Aβ levels fell through the night (see image below). In people who stayed awake, total Aβ rose 30 percent above baseline, in keeping with previous reports of increased Aβ in sleep-deprived people.
However, the percentage of 13C-labeled Aβ throughout the measurement period did not differ between sleep conditions, as evidenced by coincident SILK curves (see image below). This suggests that overall clearance occurred at the same pace in all volunteers, regardless of their sleep quality, write the authors, who base their conclusion on previous modelling of SILK kinetics (Elbert et al., 2014). Hence, they reasoned, the uptick in total Aβ in those who missed sleep must come from more synthesis.
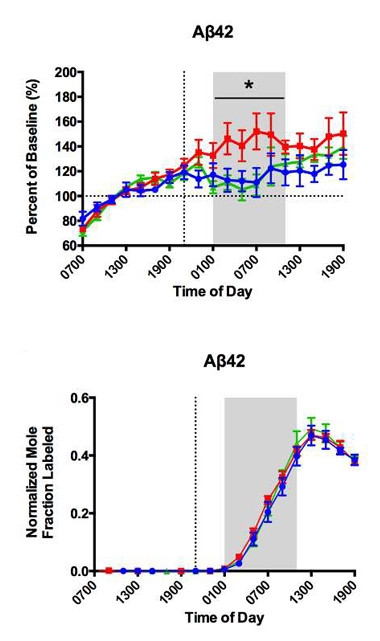
Nighttime Aβ.
Total CSF Aβ (top) waned throughout the night in people who slept normally (blue) or who took a sleep aid (green), but rose 30 percent in those who stayed awake (red). In SILK curves (bottom), the ratio of C13:C12 Aβ (bottom) rose and fell equally in all subjects. [Image courtesy of Brendan Lucey, Annals of Neurology.]
What could cause Aβ synthesis to speed up in a sleep-deprived person? Lucey said that, quite simply, increased neuronal activity is likely to blame. Neuronal activity has been linked previously to Aβ release into the interstitial fluid, which equilibrates with the CSF (Dec 2005 news; May 2011 news).
Lucey was surprised that sleep deprivation didn’t affect clearance in this study, as other groups had found sleep to be necessary for glymphatic clearance of brain waste including, presumably, Aβ (Oct 2013 news on Xie et al., 2013). This pilot study does not explain why degradation of Aβ remains steady even if a person doesn’t sleep for one night, he said.
Jeffrey Iliff, Oregon Health & Science University, Portland, said this study took a step in the direction of clarifying why people who sleep poorly have higher levels of Aβ in the brain. However, he noted that measurements taken low in the spinal column, as done here, may not capture glymphatic clearance occurring up in the brain, and Lucey agreed that clearance mechanisms could still be at work. Iliff also noted that the participants who were permitted to sleep normally achieved very little slow-wave sleep, the stage associated with reduced neuronal activity. That makes it more difficult to attribute sleep-wake changes in CSF Aβ in these people to changes in neuronal activity.
Barbara Bendlin, University of Wisconsin-Madison, wrote to Alzforum that it would be interesting to see how amyloid production and clearance differ with nighttime sleep or wakefulness in older people or in those who are already accumulating brain amyloid. The WashU group previously reported that these factors affect amyloid kinetics (Patterson et al., 2015).—Gwyneth Dickey Zakaib
References
News Citations
- CSF Aβ—New Approach Shows Rapid Flux, May Help Evaluate Therapeutics
- Paper Alert: Synaptic Activity Increases Aβ Release
- Do Overactive Brain Networks Broadcast Alzheimer’s Pathology?
- From ApoE to Zzz’s—Does Sleep Quality Affect Dementia Risk?
Paper Citations
- Yaffe K, Laffan AM, Harrison SL, Redline S, Spira AP, Ensrud KE, Ancoli-Israel S, Stone KL. Sleep-disordered breathing, hypoxia, and risk of mild cognitive impairment and dementia in older women. JAMA. 2011 Aug 10;306(6):613-9. PubMed.
- Tranah GJ, Blackwell T, Stone KL, Ancoli-Israel S, Paudel ML, Ensrud KE, Cauley JA, Redline S, Hillier TA, Cummings SR, Yaffe K, . Circadian activity rhythms and risk of incident dementia and mild cognitive impairment in older women. Ann Neurol. 2011 Nov;70(5):722-32. PubMed.
- Ooms S, Overeem S, Besse K, Rikkert MO, Verbeek M, Claassen JA. Effect of 1 night of total sleep deprivation on cerebrospinal fluid β-amyloid 42 in healthy middle-aged men: a randomized clinical trial. JAMA Neurol. 2014 Aug;71(8):971-7. PubMed.
- Ju YS, Ooms SJ, Sutphen C, Macauley SL, Zangrilli MA, Jerome G, Fagan AM, Mignot E, Zempel JM, Claassen JA, Holtzman DM. Slow wave sleep disruption increases cerebrospinal fluid amyloid-β levels. Brain. 2017 Aug 1;140(8):2104-2111. PubMed.
- Elbert DL, Patterson BW, Bateman RJ. Analysis of a compartmental model of amyloid beta production, irreversible loss and exchange in humans. Math Biosci. 2015 Mar;261:48-61. Epub 2014 Dec 9 PubMed.
- Xie L, Kang H, Xu Q, Chen MJ, Liao Y, Thiyagarajan M, O'Donnell J, Christensen DJ, Nicholson C, Iliff JJ, Takano T, Deane R, Nedergaard M. Sleep drives metabolite clearance from the adult brain. Science. 2013 Oct 18;342(6156):373-7. PubMed.
- Patterson BW, Elbert DL, Mawuenyega KG, Kasten T, Ovod V, Ma S, Xiong C, Chott R, Yarasheski K, Sigurdson W, Zhang L, Goate A, Benzinger T, Morris JC, Holtzman D, Bateman RJ. Age and amyloid effects on human central nervous system amyloid-beta kinetics. Ann Neurol. 2015 Sep;78(3):439-53. Epub 2015 Jul 20 PubMed.
Further Reading
Papers
- Lucey BP, Mawuenyega KG, Patterson BW, Elbert DL, Ovod V, Kasten T, Morris JC, Bateman RJ. Associations Between β-Amyloid Kinetics and the β-Amyloid Diurnal Pattern in the Central Nervous System. JAMA Neurol. 2017 Feb 1;74(2):207-215. PubMed.
- Chen DW, Wang J, Zhang LL, Wang YJ, Gao CY. Cerebrospinal Fluid Amyloid-β Levels are Increased in Patients with Insomnia. J Alzheimers Dis. 2018;61(2):645-651. PubMed.
- Minakawa EN, Miyazaki K, Maruo K, Yagihara H, Fujita H, Wada K, Nagai Y. Chronic sleep fragmentation exacerbates amyloid β deposition in Alzheimer's disease model mice. Neurosci Lett. 2017 Jul 13;653:362-369. Epub 2017 May 26 PubMed.
- Boespflug EL, Iliff JJ. The Emerging Relationship Between Interstitial Fluid-Cerebrospinal Fluid Exchange, Amyloid-β, and Sleep. Biol Psychiatry. 2018 Feb 15;83(4):328-336. Epub 2017 Dec 7 PubMed.
Primary Papers
- Lucey BP, Hicks TJ, McLeland JS, Toedebusch CD, Boyd J, Elbert DL, Patterson BW, Baty J, Morris JC, Ovod V, Mawuenyega KG, Bateman RJ. Effect of sleep on overnight cerebrospinal fluid amyloid β kinetics. Ann Neurol. 2018 Jan;83(1):197-204. PubMed.
Annotate
To make an annotation you must Login or Register.
Comments
University of Wisonsin
This is a fascinating new study from WashU that adds to a growing body of literature indicating that sleep disruption is linked to amyloid. Sleep has been linked to features of Alzheimer’s disease in a number of studies—both rodent and human—but an outstanding question that has been difficult to address (especially in humans) is whether disrupted sleep contributes to brain amyloid accumulation via increased amyloid production, decreased clearance, or both.
This study is really well done. Not only are amyloid levels monitored over an extended period of time using a lumbar catheter in sleep-deprived and non-deprived participants, but the investigators use a method developed by their group (Bateman et al., 2006) to quantify the production and clearance rates of CNS proteins.
Comparing sleep-deprived to non-deprived participants, the investigators found increased amyloid production in the sleep-deprived group. Markers of amyloid in CSF increased by up to 30 percent, which is substantial. Importantly, their modeling technique suggested no group differences in clearance. Given that higher amyloid concentration is associated with greater brain amyloid accumulation, the obvious question that should be addressed next is whether treating sleep disorders can reduce risk for AD by lowering amyloid concentrations.
Participants in this study were cognitively healthy, were not likely accumulating brain amyloid, and overall were fairly young. It would be interesting to see how sleep/wake amyloid kinetics differ in older age, or in participants who are already accumulating brain amyloid, given that these factors were shown in a previous study from this group to affect amyloid kinetics.
As an aside, I think the study is also relevant to the interpretation of CSF biomarkers in studies that are not focused on sleep, since we typically do not characterize sleep quality in participants who undergo lumbar puncture for research studies on AD. One way to guard against misinterpretation of high Aβ42 levels (brought on by a prior night of poor sleep), is to use Aβ42/ Aβ40 ratios.
References:
Bateman RJ, Munsell LY, Morris JC, Swarm R, Yarasheski KE, Holtzman DM. Human amyloid-beta synthesis and clearance rates as measured in cerebrospinal fluid in vivo. Nat Med. 2006 Jul;12(7):856-61. PubMed.
Radboud University Nijmegen Medical Center
I have read this pilot study performed by Lucey and colleagues with much interest. It is a carefully performed physiological study that, despite its small sample size, provides exciting new insights in the relationship between sleep and Alzheimer's disease.
In 2014, we published the results of what I think was the first attempt to translate mouse models of sleep deprivation to human physiology. We succeeded in sleep depriving 13 healthy middle-aged men during one night, with an equal-sized control group, using an indwelling catheter to sample cerebrospinal fluid levels of Aβ (40 and 42) and tau. We found that on average, CSF Aβ42 decreased (by 10 percent) after normal sleep, but remained stable after sleep deprivation. This suggested that, like in mice, sleep deprivation may increase Aβ levels in humans, and we, like others in this field had already done, postulated that repeated episodes of sleep deprivation could contribute to gradually increasing levels of Aβ, which in turn may initiate or aggravate Alzheimer pathology. It also appeared that it was the loss of slow-wave sleep that resulted mostly in the increase in amyloid levels, however, our study was not powered to prove that. Important other questions that remained were whether the increase in amyloid levels was due to increased production or to reduced clearance.
This study by Lucey et al. makes several important points. First, it replicates, in a new population, the observation that sleep deprivation affects amyloid levels in healthy humans. Second, it adds important information on amyloid production, using a laborious but precise method developed in Bateman's lab (SILK), showing that at least in some subjects, sleep deprivation leads to a strong increase in amyloid production. A strong point is that more than one amyloid species was measured, from which we learn that the increase in amyloid appears nonspecific to Aβ42. In our study we found changes in Aβ42 but not 40. This difference may be due to different assays, and I think most evidence now points toward an increase in both 40 and 42 (and 38). Third, this study finds no evidence of altered clearance processes related to sleep, indicating that the changes in amyloid levels are related to production and not clearance. In addition to the arguments provided by the authors (which mainly concern the pharmacodynamics of the SILK method) I also note that the study, which elegantly included a very strong increase in slow-wave sleep in one arm, was designed so that it could have picked up the hypothesized effects of slow-wave sleep on clearance. In a recent study from the same department (Yu, et al., 2017), specific inhibition of slow-wave sleep was applied, leading to increases in CSF amyloid levels, but not in other CSF proteins that would have been expected to be affected by clearance. Together, these studies, along with our 2014 study, replicate in humans the effects of sleep deprivation on CSF amyloid levels, but fail to replicate the animal studies suggesting a “glymphatic” clearance mechanism for amyloid.
Of course, sample size is small (but do not underestimate the tremendous complexity of this study), however, physiological studies can provide reliable and relevant insights even with small sample sizes. Also, inter-individual variability is strong, and will likely be a topic for further investigation. These arguments however, in my opinion, should cast no doubts on the relevance of this pilot study.
References:
Ju YS, Ooms SJ, Sutphen C, Macauley SL, Zangrilli MA, Jerome G, Fagan AM, Mignot E, Zempel JM, Claassen JA, Holtzman DM. Slow wave sleep disruption increases cerebrospinal fluid amyloid-β levels. Brain. 2017 Aug 1;140(8):2104-2111. PubMed.
Make a Comment
To make a comment you must login or register.