ApoE4 Promotes Amyloidosis, But Only in Plaque-Free Mice
Quick Links
The ApoE4 allele boosts the risk for sporadic AD by promoting amyloid deposition, but at what stage of amyloidosis does the gene exert its greatest effect? Two papers in the December 6 Neuron offer the same answer—that ApoE4 facilitates Aβ fibrillization and plaque seeding, but that it has little effect once plaques are established. Researchers led by David Holtzman at Washington University School of Medicine in St. Louis arrived at this conclusion by suppressing human ApoE3 or ApoE4 in AD mice either from birth or after plaques emerged. Conversely, researchers led by Guojun Bu at the Mayo Clinic in Jacksonville, Florida, overexpressed human ApoE3 or ApoE4 in AD mouse models at different ages. In both cases, the intervention only affected plaque load when it began before plaques emerged. How ApoE stimulates plaque formation, and whether that involves a direct interaction with Aβ, remains unclear.
- The ApoE4 allele affected amyloidosis during initial plaque seeding.
- Once plaques appeared, changing ApoE4 did not budge amyloid load.
- However, ApoE4 still exacerbated toxicity in the amyloid-laden brain.
“Strikingly, the two studies arrive at identical conclusions by using different molecular genetic approaches to move ApoE levels in opposite directions. … The concordance of results obtained by these different approaches strongly supports the role of ApoE in Aβ seed formation rather than plaque growth,” wrote Robert Vassar at Northwestern University, Chicago, in an accompanying editorial.
Others called the findings significant. “These are important papers and will be highly cited in the future for [helping the field] understand the role of ApoE in promoting pathogenesis,” predicted William Rebeck at Georgetown University, Washington D.C.
Suppressing ApoE4 Protects Against Amyloidosis and Neuron Damage
Numerous studies of mice and people have found that the ApoE4 allele revs up amyloid deposition and leads to earlier onset of amyloidosis (Apr 2009 news; Bales et al., 2009; Jansen et al., 2015). However, amyloidosis in the brain is not constant, but instead follows a sigmoid curve that begins with a slow nucleation phase that gives way to a period of rapid growth. Might ApoE have a greater effect at one of these phases?
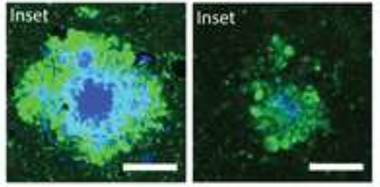
Less Toxic Plaques.
When ApoE4 expression was suppressed in plaque-laden mice (right), plaques contained less fibrillar material (blue) 10 weeks later, and accumulated fewer dystrophic neurites (green) than in controls that continuously expressed the apolipoprotein (left). [Courtesy of Huynh et al., Neuron.]
To find out, Holtzman and colleagues took knock-in mice that express human ApoE3 or ApoE4, but no endogenous mouse ApoE, and crossed them to APPPS1-21 mice, which begin to develop plaques in the cortex as early as six weeks of age. First author Tien-Phat Huynh administered antisense oligonucleotides (ASOs) against ApoE to the offspring either at birth or at six weeks. The authors gave one booster dose a few weeks later to keep expression low. When assessed one or two months after the injection, the ASOs had knocked down ApoE mRNA and protein levels by about 50 percent.
When Huynh assessed the mice at 16 weeks old, those with ApoE4 suppressed from birth had about half as much plaque in the cortex as controls. Levels of soluble and insoluble Aβ40 and Aβ42 in the cortex dropped as well, in keeping with a previous study that found ApoE boosts transcription of the amyloid precursor protein (Jan 2017 news). Notably, the mice also developed about half as many dystrophic neurites, indicating less neuronal damage than in controls with normal ApoE4 transgene expression. Suppressing the ApoE3 transgene from birth, on the other hand, only slightly lowered fibrillar plaque and insoluble Aβ, and did not change soluble Aβ, suggesting ApoE3 has a weaker effect on amyloid pathology.
These findings contrasted with the outcome when ApoE was first suppressed at six weeks, i.e., after amyloidosis had already begun. When these animals were 16 weeks old, fibrillar plaque load and total Aβ level were as rampant as in controls. However, suppression of ApoE4 did cause some subtle effects. Individual plaques were larger in area and more diffuse, containing more non-fibrillar material than in controls. Surprisingly, despite the minimal effect on plaques, the amount of dystrophic neurites dropped by half, just as it did in the mice treated from birth (see image above). Soluble Aβ42 nudged up compared to controls, while insoluble Aβ40 dropped slightly, suggesting subtle changes in different pools of the peptide, the authors noted. The reason for these variations was unclear.
What might explain the changes in plaques? Previous work has found that in the complete absence of ApoE, Aβ still accumulates, but virtually none of it becomes fibrillar (Bales et al., 1997). “ApoE is required for the fibrillization of Aβ in vivo. By lowering ApoE, we’re getting plaques that are less fibrillar and larger,” Holtzman explained. Bu noted that these plaques seem to be less damaging to surrounding tissue, based on the dearth of dystrophic neurites. “In the absence of ApoE, amyloid is not as toxic,” Bu told Alzforum.
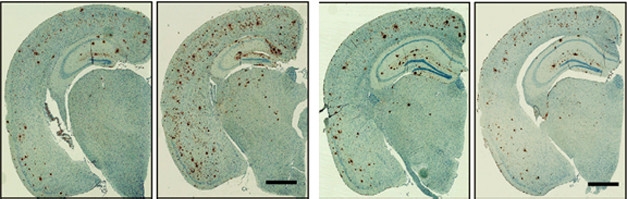
Early effects. Expressing human ApoE4 from birth (panel 2) doubled the plaque load (brown) at nine months as compared to that in mice without human ApoE (left). Turning on ApoE4 after plaques had formed (last panel) had no effect compared to controls without ApoE4 (panel 3). [Courtesy of Liu et al., Neuron.]
ApoE4 Overexpression Burgeons Plaque Load
Bu and colleagues took the opposite approach from Holtzman, overexpressing ApoE, and their findings dovetailed almost perfectly. To selectively boost ApoE in the brain at specific times, the researchers first generated inducible ApoE mouse models. First author Chia-Chen (Jenny) Liu inserted transgenes carrying human ApoE3 or ApoE4 into the mouse genome. A stop codon kept the transgenes inactive except in cells carrying Cre recombinase, which snipped out the stop signal. Liu and colleagues crossed these mice to a strain that expressed Cre recombinase in astrocytes, resulting in expression of the transgenes only in those brain cells. Astrocytes are the major source of ApoE in the CNS. In the ApoE/Cre crosses, astrocytes pumped out enough of the human protein to reach two to three times the level seen in control mice that constitutively express human ApoE. In addition, the transgenes contained an antibiotic response element that allowed them to be switched off by feeding the mice doxycycline for two weeks.
The authors then crossed these mice to an APP/PSI strain that develops plaques at about four months of age. In one group of offspring, the researchers allowed the ApoE transgenes to stay on from birth to nine months of age. In a second group, they turned the transgenes off at six months, and in a third group, they kept the transgenes off for the first six months and then turned them on for the next three.
The results were dramatically different. When ApoE4 was active during the first six months of life, levels of soluble and insoluble Aβ40 and Aβ42 as well as Aβ oligomers more than doubled by nine months, and the number of plaques mushroomed 1.5- to twofold compared with control mice without human ApoE (see image above). On the other hand, turning on ApoE4 only from six to nine months had no effect on any form of Aβ or plaque load.
ApoE3 expression did not affect amyloid at either age, in contrast to Holtzman’s findings that E3 mildly promoted amyloidosis. Steven Paul, from the biotech company Voyager Therapeutics in Cambridge, Massachusetts, suggested that the presence of endogenous mouse ApoE in the overexpression models could confound the ApoE3 results. Because mouse ApoE also stimulates amyloidosis, the ApoE3 allele might provide no additional impetus to the process, Paul speculated. Bu is repeating these studies on a mouse ApoE knockout background.
As in Holtzman’s study, ApoE4 level also affected neuron damage and inflammation. Compared with controls, mice that expressed ApoE4 for the first six months of life developed more than twice as many dystrophic neurites. They also harbored more activated astrocytes and microglia, as well as higher levels of pro-inflammatory cytokines. However, turning on ApoE4 late had no effect on dystrophic neurites or inflammation. Intriguingly, the ApoE3 allele lowered astrogliosis and pro-inflammatory cytokines and nudged up levels of the postsynaptic marker PSD95, hinting at beneficial effects on synapses.
Other researchers said Bu’s inducible model system will aid future ApoE research. “This is an elegant and technically well-done study,” Paul said. Rebeck agreed that Bu has versatile models, and noted that the ability to express ApoE only in the brain avoids confounding effects from the periphery. Bu plans to express ApoE4 only in microglia, the other brain cell that produces the protein. In other experiments, he will examine the effect of ApoE in circulation by expressing the protein only in the liver (Oct 2016 news). Paul suggested expressing ApoE2 in the brain to explore whether that protective allele lowers amyloidosis.
What’s ApoE Doing?
These studies do not address how ApoE4 facilitates plaque formation, the researchers agreed. Previous studies have found that the allele triggers oligomerization of Aβ and also slows its clearance from the brain (Jul 2010 conference news; Jul 2011 webinar; Oct 2012 news). Both of these mechanisms might be at work in the inducible ApoE mice, Bu said. In the current study, he found that expression of ApoE4, but not ApoE3, in the young mice nearly doubled the half-life of Aβ40 in the interstitial fluid, suggesting slower clearance that would raise local concentrations of Aβ, promoting its aggregation. The higher expression of soluble Aβ42 in the presence of ApoE4 would also contribute to this process. In addition, ApoE4 may directly interact with Aβ42 to change its shape and spur fibrillization. The E4 allele is more unstructured than E3 and aggregates more easily, Bu noted.
The researchers stressed that ApoE may act through amyloid-independent processes as well, such as insulin signaling or inflammation (Oct 2017 news). “Even around fibrillar plaques, there was less neuritic damage when we lowered ApoE. ApoE may influence the injury response in the setting of a toxic stimulus,” Holtzman suggested. In keeping with this idea, his group recently reported that ApoE4 exacerbates tau pathology independently of its effects on Aβ (Sep 2017 news). In future studies, it will be important to modulate ApoE4 in mouse models that develop both amyloid and tau pathology, in order to better predict what might happen in AD, Holtzman noted.
How relevant are these findings for human disease? The apparent role of ApoE4 in promoting plaque formation fits well with human data, in which carriers have an earlier age of disease onset, researchers said (Ossenkoppele et al., 2015; Gonneaud et al., 2016). Thus, therapies directed against ApoE4 might work best in a preventive paradigm, Rebeck said. Others agreed, but noted that the mouse data suggest late treatment might still help lower the toxicity of Aβ and benefit the brain.
Some groups are working on anti-ApoE4 therapies. Voyager Therapeutics uses RNAi to silence the E4 allele in mouse models. ASOs targeting other genes are currently in clinical trials for several neurological disorders, and have been approved for spinal muscular atrophy (Nov 2016 news).—Madolyn Bowman Rogers
References
News Citations
- More ApoE4 Means More Amyloid in Brains of Middle-Aged
- ApoE Risk Explained? Isoform-Dependent Boost in APP Expression Uncovered
- Distinct Roles for Brain and Blood ApoE in Neuron Health?
- St. Louis: ApoE—A Clearer View of its Role In AD?
- ApoE4 Promotes Aβ Oligomerization
- ApoE4 Traps Insulin Receptor Inside Neurons
- ApoE4 Makes All Things Tau Worse, From Beginning to End
- Positive Trials of Spinal Muscular Atrophy Bode Well for Antisense Approach
Research Models Citations
Webinar Citations
Paper Citations
- Bales KR, Liu F, Wu S, Lin S, Koger D, Delong C, Hansen JC, Sullivan PM, Paul SM. Human APOE isoform-dependent effects on brain beta-amyloid levels in PDAPP transgenic mice. J Neurosci. 2009 May 27;29(21):6771-9. PubMed.
- Jansen WJ, Ossenkoppele R, Knol DL, Tijms BM, Scheltens P, Verhey FR, Visser PJ, Amyloid Biomarker Study Group, Aalten P, Aarsland D, Alcolea D, Alexander M, Almdahl IS, Arnold SE, Baldeiras I, Barthel H, van Berckel BN, Bibeau K, Blennow K, Brooks DJ, van Buchem MA, Camus V, Cavedo E, Chen K, Chetelat G, Cohen AD, Drzezga A, Engelborghs S, Fagan AM, Fladby T, Fleisher AS, van der Flier WM, Ford L, Förster S, Fortea J, Foskett N, Frederiksen KS, Freund-Levi Y, Frisoni GB, Froelich L, Gabryelewicz T, Gill KD, Gkatzima O, Gómez-Tortosa E, Gordon MF, Grimmer T, Hampel H, Hausner L, Hellwig S, Herukka SK, Hildebrandt H, Ishihara L, Ivanoiu A, Jagust WJ, Johannsen P, Kandimalla R, Kapaki E, Klimkowicz-Mrowiec A, Klunk WE, Köhler S, Koglin N, Kornhuber J, Kramberger MG, Van Laere K, Landau SM, Lee DY, de Leon M, Lisetti V, Lleó A, Madsen K, Maier W, Marcusson J, Mattsson N, de Mendonça A, Meulenbroek O, Meyer PT, Mintun MA, Mok V, Molinuevo JL, Møllergård HM, Morris JC, Mroczko B, Van der Mussele S, Na DL, Newberg A, Nordberg A, Nordlund A, Novak GP, Paraskevas GP, Parnetti L, Perera G, Peters O, Popp J, Prabhakar S, Rabinovici GD, Ramakers IH, Rami L, Resende de Oliveira C, Rinne JO, Rodrigue KM, Rodríguez-Rodríguez E, Roe CM, Rot U, Rowe CC, Rüther E, Sabri O, Sanchez-Juan P, Santana I, Sarazin M, Schröder J, Schütte C, Seo SW, Soetewey F, Soininen H, Spiru L, Struyfs H, Teunissen CE, Tsolaki M, Vandenberghe R, Verbeek MM, Villemagne VL, Vos SJ, van Waalwijk van Doorn LJ, Waldemar G, Wallin A, Wallin ÅK, Wiltfang J, Wolk DA, Zboch M, Zetterberg H. Prevalence of cerebral amyloid pathology in persons without dementia: a meta-analysis. JAMA. 2015 May 19;313(19):1924-38. PubMed.
- Bales KR, Verina T, Dodel RC, Du Y, Altstiel L, Bender M, Hyslop P, Johnstone EM, Little SP, Cummins DJ, Piccardo P, Ghetti B, Paul SM. Lack of apolipoprotein E dramatically reduces amyloid beta-peptide deposition. Nat Genet. 1997 Nov;17(3):263-4. PubMed.
- Ossenkoppele R, Jansen WJ, Rabinovici GD, Knol DL, van der Flier WM, van Berckel BN, Scheltens P, Visser PJ, Amyloid PET Study Group, Verfaillie SC, Zwan MD, Adriaanse SM, Lammertsma AA, Barkhof F, Jagust WJ, Miller BL, Rosen HJ, Landau SM, Villemagne VL, Rowe CC, Lee DY, Na DL, Seo SW, Sarazin M, Roe CM, Sabri O, Barthel H, Koglin N, Hodges J, Leyton CE, Vandenberghe R, van Laere K, Drzezga A, Forster S, Grimmer T, Sánchez-Juan P, Carril JM, Mok V, Camus V, Klunk WE, Cohen AD, Meyer PT, Hellwig S, Newberg A, Frederiksen KS, Fleisher AS, Mintun MA, Wolk DA, Nordberg A, Rinne JO, Chételat G, Lleo A, Blesa R, Fortea J, Madsen K, Rodrigue KM, Brooks DJ. Prevalence of amyloid PET positivity in dementia syndromes: a meta-analysis. JAMA. 2015 May 19;313(19):1939-49. PubMed.
- Gonneaud J, Arenaza-Urquijo EM, Fouquet M, Perrotin A, Fradin S, de La Sayette V, Eustache F, Chételat G. Relative effect of APOE ε4 on neuroimaging biomarker changes across the lifespan. Neurology. 2016 Oct 18;87(16):1696-1703. Epub 2016 Sep 28 PubMed.
External Citations
Further Reading
News
- ApoE Does Not Bind Aβ, Competes for Clearance
- Has ApoE’s Time Come as a Therapeutic Target?
- ApoE Risk Explained? Isoform-Dependent Boost in APP Expression Uncovered
- ApoE Variants Modulate Astrocyte Appetite for Synapses
- New Look at Sex and ApoE4 Puts Women at Risk Earlier than Men
- APOE4 Subtly Alters Brain Network Activity With Age
- Lowering ApoE Brings Down Amyloid in Mice
Primary Papers
- Liu CC, Zhao N, Fu Y, Wang N, Linares C, Tsai CW, Bu G. ApoE4 Accelerates Early Seeding of Amyloid Pathology. Neuron. 2017 Dec 6;96(5):1024-1032.e3. PubMed.
- Huynh TV, Liao F, Francis CM, Robinson GO, Serrano JR, Jiang H, Roh J, Finn MB, Sullivan PM, Esparza TJ, Stewart FR, Mahan TE, Ulrich JD, Cole T, Holtzman DM. Age-Dependent Effects of apoE Reduction Using Antisense Oligonucleotides in a Model of β-amyloidosis. Neuron. 2017 Dec 6;96(5):1013-1023.e4. PubMed.
- Vassar R. Seeds of Destruction: New Mechanistic Insights into the Role of Apolipoprotein E4 in Alzheimer's Disease. Neuron. 2017 Dec 6;96(5):953-955. PubMed.
Annotate
To make an annotation you must Login or Register.
Comments
No Available Comments
Make a Comment
To make a comment you must login or register.