Unexpected Polypeptides Pop Up in Huntington’s Disease
Quick Links
Scientists have been missing out on four repeat polypeptides in Huntington’s disease, according to new data from the lab of Laura Ranum at the University of Florida in Gainesville. She discovered that the same trinucleotide repeats that encode well-known polyglutamine tracts in huntingtin can also be translated, sans start codon, into polyserine, polyalanine, polyleucine, and polycysteine. Moreover, the novel polypeptides appear alongside the polyglutamine-expanded huntingtin, and sometimes even overshadow it, in the parts of the brain that degenerate in HD. Ranum presented these results at RNA Metabolism in Neurological Disease, a satellite symposium to the Society for Neuroscience annual meeting, held October 15-16 in Chicago. Several scientists in attendance called Ranum’s news one of the highlights of the conference. The work also appears in the November 18 Neuron.
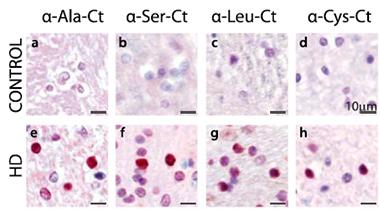
Polypeptide variety.
The striata of people who died of HD, but not controls, contained four RAN polypeptides. [Courtesy of Neuron, Bañez-Coronel et al.]
Previously, Ranum discovered that repetitive gene sequences sometimes get processed through what is known as repeat-associated non-ATG (RAN) translation (Zu et al., 2011). For example, hexanucleotide repeats in the C9ORF72 gene, linked to both amyotrophic lateral sclerosis and frontotemporal dementia, are copied and translated in both the sense and antisense directions, yielding a set of poly-dipeptides that scientists suspect are toxic (see Nov 2013 news). However, those repeats—and every other sequence observed to undergo RAN translation—reside in an intron. Could the same thing happen in coding repeats?
To find out, first author Monica Bañez-Coronel and colleagues picked the huntingtin gene as a possible candidate because it contains a stretch of CAG codons. People with 36 or more of these repeats are at risk for HD, and those with 40 or more are almost certain to develop the disease. Upward of 50 repeats, carriers are likely to develop HD early in life, some even before adulthood. If those CAG codons were to undergo RAN translation in the sense direction, they could produce polyserines and polyalanines, in addition to the canonical polyglutamine. The antisense strand would yield polyleucine, polycysteine, and polyalanine. The authors wanted to generate antibodies to each, but it can be difficult to make antibodies to repetitive peptides, Ranum said.
Fortunately, each potential polypeptide would be followed by a unique amino acid sequence as translation continued beyond the repeats until it hit a stop codon. Bañez-Coronel generated antibodies to four of those post-repeat peptides. (The strategy did not work for polyalanine because the stop codon occurred right after the repeat). She applied these antibodies to autopsy samples taken from the striatum, a brain region usually affected in HD. All four lit up neurons, astrocytes, and microglia in brains from people with Huntington’s, but not controls (see image above). The polypeptides were found in the nucleus and cytoplasm, and appeared diffuse and in punctate aggregates.
Bañez-Coronel compared immunohistochemistry for polyglutamine-expanded huntingtin to that of the RAN-translated poly amino acids in different subregions of the striatum. Notably, in the caudate and putamen, she found more cells positive for RAN polypeptides than for polyglutamine. In the white-matter tracts associated with those regions she saw intense RAN protein signals, whereas polyglutamine did not appear at all.
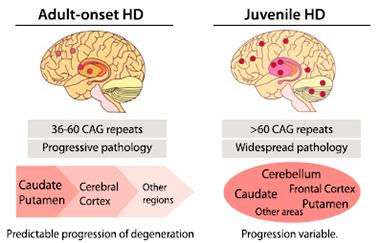
Step by step.
Neurodegeneration progresses predictably in people with adult-onset disease, but varies in those with juvenile-onset disorder. [Courtesy of Neuron, Bañez-Coronel et al.]
The striatal regions of the brain are typically the first to degenerate in HD, followed by the cortex. In the adult tissues, Bañez-Coronel detected RAN polypeptides in the cortex and cerebellum as well, though the antibody signal was less intense than in the striatum. Polyglutamine, in contrast, occurred in the cortex but never reached the cerebellum.
In juvenile-onset cases, progression of pathology varies (see image at left). The authors wondered if RAN polypeptides might help explain that variation. They examined brains from five juvenile cases. All expressed RAN proteins in the striatum, frontal cortex, and cerebellum. In the two brains with the most severe cerebellar atrophy, RAN peptides, but not polyglutamine, appeared in the cerebellum, Ranum said.
In addition to its variable sites of pathology, juvenile HD typically results from longer expansions in the Htt gene. To find out how repeat length affects RAN translation, the authors transfected HEK293T cells with minigenes containing huntingtin’s first exon and 23-80 repeats. They focused on the sense polypeptides containing alanine or serine, since they were comparing them to the polyglutamine-containing huntingtin fragment produced by normal, ATG-associated translation in the sense direction. These minigenes always produced polyglutamine-htt regardless of repeat size. They made polyserine if the repeat length was 35 or more, and polyalanine when the extension grew to 50. In addition, the serine polypeptide started to aggregate once it reached about 45 amino acids long. Thus, the polyalanine and insoluble polyserine associated with longer repeat lengths.
Dieter Edbauer of Ludwig-Maximilians University of Munich, who did not participate in the work, told Alzforum he was particularly intrigued by the results with juvenile HD. Carriers of longer repeats might get sick because of more RAN translation, rather than simply a longer polyglutamine stretch within the huntingtin protein, he said. And the anatomical distribution of those RAN polypeptides differs from the polyglutamine-expanded huntingtin, potentially explaining why different regions are affected in juvenile and adult-onset HD.
Supporting this theory, the authors saw indicators that the RAN polypeptides could be toxic. The brain regions where RAN products were prevalent, the caudate and the putamen and their white-matter bundles, tested positive for Iba1, a microglial marker of neuroinflammation. Those spots also tested positive for caspase 3, a marker of apoptosis, indicating that those neurons and glia with RAN polypeptides were sick or dying. Bañez-Coronel confirmed that RAN polypeptides were toxic when she transfected minigenes into neuronal and glial cell cultures.
“These new mutant proteins may contribute to the disease,” Ranum concluded. At this point, she added, she cannot be sure about the relevant contributions of polyglutamine versus the RAN polypeptides. One implication, the authors noted, is that researchers trying to treat Huntington’s may have to expand their ideas to interfere with RAN translation in both the sense and antisense directions.
“The finding of intense RAN protein accumulation in white-matter regions in the absence of significant polyglutamine accumulation is particularly interesting,” Bañez-Coronel told Alzforum. “White-matter changes can occur early in disease and are also detected in presymptomatic patients. Our findings suggest that RAN proteins are likely contributing to these white-matter alterations.”
In addition, Ranum pointed out that several other neurological diseases, including spinocerebellar ataxia and spinal-bulbar muscular atrophy, are associated with polyglutamine stretches in coding regions. She speculated that RAN translation might occur in those conditions as well. Tom Cooper of the Baylor College of Medicine, Houston, told Alzforum that Ranum’s work convinced him to be on the lookout for RAN translation. He will be checking his mice modeling myotonic dystrophy type 1 for signs of RAN polypeptides.—Amber Dance
References
News Citations
Paper Citations
- Zu T, Gibbens B, Doty NS, Gomes-Pereira M, Huguet A, Stone MD, Margolis J, Peterson M, Markowski TW, Ingram MA, Nan Z, Forster C, Low WC, Schoser B, Somia NV, Clark HB, Schmechel S, Bitterman PB, Gourdon G, Swanson MS, Moseley M, Ranum LP. Non-ATG-initiated translation directed by microsatellite expansions. Proc Natl Acad Sci U S A. 2011 Jan 4;108(1):260-5. Epub 2010 Dec 20 PubMed.
Further Reading
Papers
- Yang D, Abdallah A, Li Z, Lu Y, Almeida S, Gao FB. FTD/ALS-associated poly(GR) protein impairs the Notch pathway and is recruited by poly(GA) into cytoplasmic inclusions. Acta Neuropathol. 2015 Oct;130(4):525-35. Epub 2015 Jun 2 PubMed.
- Gendron TF, van Blitterswijk M, Bieniek KF, Daughrity LM, Jiang J, Rush BK, Pedraza O, Lucas JA, Murray ME, Desaro P, Robertson A, Overstreet K, Thomas CS, Crook JE, Castanedes-Casey M, Rousseau L, Josephs KA, Parisi JE, Knopman DS, Petersen RC, Boeve BF, Graff-Radford NR, Rademakers R, Lagier-Tourenne C, Edbauer D, Cleveland DW, Dickson DW, Petrucelli L, Boylan KB. Cerebellar c9RAN proteins associate with clinical and neuropathological characteristics of C9ORF72 repeat expansion carriers. Acta Neuropathol. 2015 Oct;130(4):559-73. Epub 2015 Sep 8 PubMed.
- Oh SY, He F, Krans A, Frazer M, Taylor JP, Paulson HL, Todd PK. RAN translation at CGG repeats induces ubiquitin proteasome system impairment in models of fragile X-associated tremor ataxia syndrome. Hum Mol Genet. 2015 Aug 1;24(15):4317-26. Epub 2015 May 7 PubMed.
- Kino Y, Washizu C, Kurosawa M, Oma Y, Hattori N, Ishiura S, Nukina N. Nuclear localization of MBNL1: splicing-mediated autoregulation and repression of repeat-derived aberrant proteins. Hum Mol Genet. 2015 Feb 1;24(3):740-56. Epub 2014 Sep 30 PubMed.
- Wojciechowska M, Olejniczak M, Galka-Marciniak P, Jazurek M, Krzyzosiak WJ. RAN translation and frameshifting as translational challenges at simple repeats of human neurodegenerative disorders. Nucleic Acids Res. 2014 Oct 29;42(19):11849-64. Epub 2014 Sep 12 PubMed.
Primary Papers
- Bañez-Coronel M, Ayhan F, Tarabochia AD, Zu T, Perez BA, Tusi SK, Pletnikova O, Borchelt DR, Ross CA, Margolis RL, Yachnis AT, Troncoso JC, Ranum LP. RAN Translation in Huntington Disease. Neuron. 2015 Nov 18;88(4):667-77. PubMed.
Annotate
To make an annotation you must Login or Register.
Comments
No Available Comments
Make a Comment
To make a comment you must login or register.