New Therapeutic Idol? Silencing Ligase Prevents Aβ Deposition in Mice
Quick Links
Researchers may have a new therapeutic target to worship. Getting rid of an E3 ubiquitin ligase known as Idol prevented amyloid pathology in mice, according to a study published November 18 in Science Translational Medicine. AD model mice lacking Idol had less Aβ and fewer plaques in the brain, according to the research. Because Idol normally targets the low-density lipoprotein receptor (LDLR) for destruction, levels of the receptor rose in the knockouts, which led to enhanced clearance of ApoE from the brain. The researchers, led by Peter Tontonoz at the University of California, Los Angeles, and Jungsu Kim at the Mayo Clinic in Jacksonville, Florida, speculate that removal of ApoE explains the dearth of Aβ, however, details of that interaction have yet to be ironed out.
Some commenters agreed. “The results appear strong, solid, and clear,” commented David Holtzman of Washington University in St. Louis. “If Idol can be targeted in the brain, it seems like a strong target for AD therapeutics.”
LDLR resides on the cell surface and plays a major role in lipid metabolism, as it latches onto lipoproteins such as ApoE and internalizes them (see Go and Mani, 2012). Previous work from Tontonoz’s lab revealed that LDLR expression is controlled at the protein level by Idol (aka inducible degrader of the LDLR), which is active in the intestine, adipose tissue, and macrophages, but not the liver (see Zelcer et al., 2009; Zhang et al., 2012). Whether Idol modulates LDLR in the brain was unknown, but previous studies indicated that elevating LDLR expression in the brain helped clear both ApoE and Aβ (see Dec 2009 news; Castellano et al., 2012).
Co-first authors Jinkuk Choi and Jie Gao and colleagues set out to determine if Idol could modulate LDLR (and thus ApoE and Aβ levels in the brain). They readily detected Idol mRNA in multiple regions of the mouse brain, where it was primarily expressed by neurons and microglia. Seven-month-old Idol knockout mice had more than twice as much LDLR in the brain as animals expressing the ligase, while animals expressing a single copy had an intermediate amount.
To investigate the potential role of Idol in amyloid metabolism, the researchers bred the Idol knockouts with APPPS1 mice. At seven months of age, male and female APPPS1 animals lacking Idol had lower concentrations of both soluble and insoluble Aβ40 and Aβ42 in their brain than did control APPPS1 mice. Males with one copy of Idol had intermediate levels of the peptides, but this dosage effect was absent in female mice. Plaque burden in the cortex of AD mice lacking Idol—as measured by immunohistochemistry and thioflavin S staining—was about half that of controls. Idol deficiency affected neither APP nor BACE1 expression, leading the researchers to hypothesize that loss of Idol somehow hastened the clearance of Aβ.
APPPS1 Idol knockouts had more LDLR in the brain and less ApoE, in keeping with LDLR’s role in the clearance of the apolipoprotein. Insoluble ApoE was most strongly suppressed, in agreement with previous reports that ApoE accumulates in plaques with Aβ.
How might Idol deficiency facilitate the removal of ApoE and Aβ? The researchers looked for answers in primary cultures of microglia, since these cells are known to gobble up proteins via phagocytosis. They monitored the internalization of fluorescently labeled Aβ42 peptides, finding that cells lacking Idol took in nearly twice as much peptide. Their appetite for ApoE was similarly enhanced. In both cases, uptake depended upon LDLR—silencing the receptor by siRNA suppressed microglial ingestion of both proteins.
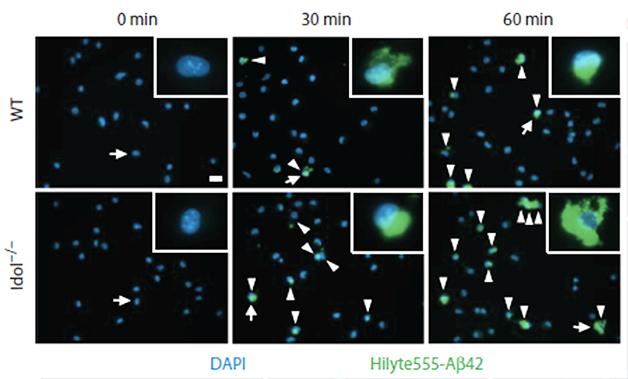
Microglial Munchies. Microglia lacking Idol (bottom) internalize more Aβ42 (green) than wild-type microglia (top). [Courtesy of Choi et al., Science Translational Medicine 2015.]
Choi and colleagues found roughly a third fewer Iba1+ microglia in the hippocampi and cortices of APPPS1 mice lacking Idol than in those expressing the ligase. These mice had about half as many activated microglia, as assessed by CD45 staining. However, the microglia appeared to be fully functional: They expressed the normal complement of microglial proteins and were roused into action by lipopolysaccharide. Tontonoz speculated that in AD mice lacking Idol, the increased expression of LDLR on microglia enhances clearance of Aβ and ApoE, which prevents the formation of plaques and thus microgliosis. The biggest unanswered question is if and how the clearance of ApoE relates to the reduction in Aβ.
“While they show that microglia lacking Idol can take up more Aβ, the study doesn't formally address whether their observed effects in vivo reflect Aβ as a ligand of the LDL receptor, or whether it is ferried into the cell through direct or indirect effects of ApoE binding to LDLR,” commented Gary Landreth of Case Western Reserve University in Cleveland. Holtzman agreed, but added that either way, the results suggest that raising LDLR levels would be a good therapeutic strategy for AD (see full comment below).
Blocking the E3 ubiquitin ligase activity of Idol could be the most straightforward way to do this. “As much as we appreciate the importance of ApoE and its receptors in brain lipid metabolism and Aβ clearance, these molecules are in general difficult to target for therapeutic intervention in AD,” wrote Guojun Bu of the Mayo Clinic in Jacksonville, Florida. “But now we have an enzymatic target that directly impacts brain metabolism of both ApoE and Aβ.”
Tontonoz added that lower Aβ loads in mice expressing only a single copy of Idol support the concept of targeting the ligase. Given that it is unlikely any inhibitor will ever block Idol entirely, it is nice to see that a 50 percent reduction can prevent plaque formation, he told Alzforum. “This dosage effect is consistent with the idea that a small molecule inhibitor could work,” he said.
Cheryl Wellington of the University of British Columbia in Vancouver pointed out that researchers will need to determine whether inhibition of Idol, and reduction in ApoE, could have other consequences besides clearing Aβ. For example, ApoE and LDLR have been implicated in neurite outgrowth (see Pitas et al., 1998; Do et al., 2015). “A closer look at neuronal Idol function, especially with respect to synaptic and behavioral effects under baseline and neuronal stress conditions, are eagerly awaited,” she wrote (see comment below).—Jessica Shugart
References
News Citations
Research Models Citations
Paper Citations
- Go GW, Mani A. Low-density lipoprotein receptor (LDLR) family orchestrates cholesterol homeostasis. Yale J Biol Med. 2012 Mar;85(1):19-28. Epub 2012 Mar 29 PubMed.
- Zelcer N, Hong C, Boyadjian R, Tontonoz P. LXR regulates cholesterol uptake through Idol-dependent ubiquitination of the LDL receptor. Science. 2009 Jul 3;325(5936):100-4. Epub 2009 Jun 11 PubMed.
- Zhang L, Reue K, Fong LG, Young SG, Tontonoz P. Feedback regulation of cholesterol uptake by the LXR-IDOL-LDLR axis. Arterioscler Thromb Vasc Biol. 2012 Nov;32(11):2541-6. Epub 2012 Aug 30 PubMed.
- Castellano JM, Deane R, Gottesdiener AJ, Verghese PB, Stewart FR, West T, Paoletti AC, Kasper TR, DeMattos RB, Zlokovic BV, Holtzman DM. Low-density lipoprotein receptor overexpression enhances the rate of brain-to-blood Aβ clearance in a mouse model of β-amyloidosis. Proc Natl Acad Sci U S A. 2012 Sep 18;109(38):15502-7. Epub 2012 Aug 27 PubMed.
- Pitas RE, Ji ZS, Weisgraber KH, Mahley RW. Role of apolipoprotein E in modulating neurite outgrowth: potential effect of intracellular apolipoprotein E. Biochem Soc Trans. 1998 May;26(2):257-62. PubMed.
- Do HT, Bruelle C, Pham DD, Jauhiainen M, Eriksson O, Korhonen LT, Lindholm D. Nerve growth factor (NGF) and pro-NGF increase low-density lipoprotein (LDL) receptors in neuronal cells partly by different mechanisms: role of LDL in neurite outgrowth. J Neurochem. 2015 Oct 20; PubMed.
Further Reading
Papers
- Hong C, Marshall SM, McDaniel AL, Graham M, Layne JD, Cai L, Scotti E, Boyadjian R, Kim J, Chamberlain BT, Tangirala RK, Jung ME, Fong L, Lee R, Young SG, Temel RE, Tontonoz P. The LXR-Idol axis differentially regulates plasma LDL levels in primates and mice. Cell Metab. 2014 Nov 4;20(5):910-8. PubMed.
Annotate
To make an annotation you must Login or Register.
Comments
Indiana University School of Medicine
This is a compelling study from the Tontonoz lab demonstrating the role of LDLR in amyloid pathogenesis in a mouse model of AD. This work follows directly from their discovery that Idol acts to promote the proteolytic degradation of LDLR through its ability to ubiquitinate the receptor. This study strongly supports and validates the hypothesis that LDLR is a quantitatively significant regulator of both ApoE and amyloid metabolism. The LDLR-dependent regulation of ApoE levels is associated with alteration of both soluble and deposited forms of Aβ. Moreover, it provides new evidence of the complexity of ApoE-dependent regulation of amyloidogensis.
The outcome of this study was not predictable given that Idol has tissue-specific actions, with robust regulation of LDLR levels in fat, but not in the liver, and had never been examined in the brain. The authors have shown high levels of Idol in microglia, with lower levels in neurons and little expression in astrocytes. The study sheds little light on the relative contribution of the various cell types on amyloid metabolism. While they show that Idol knockout microglia can take up more Aβ than wild-type microglia, the study doesn't formally address the issue of whether their observed effects in vivo reflect Aβ as a ligand of the receptor or whether Aβ is ferried into the cell through direct or indirect effects of ApoE binding to LDLR.
The Holtzman lab had previously demonstrated a remarkable effect of overexpression of LDLR on ApoE levels and this work is entirely consistent with those findings. A remarkable finding in the former study of LDLR overexpression was a large effect of gender with greater effects observed in males, however, analogous effects were not evident in the present study. A potential confound in their interpretation of the actions of Idol and LDLR is that Idol also targets VLDLR and ApoER2, but given the similarity of the experimental outcomes they are probably correct in arguing that LDLR is the dominant receptor in play.
Overall, this is an important contribution to understanding LDLR and ApoE actions in AD pathogenesis.
University of Pittsburgh
In the study by Choi et al., the authors test the effect of Idol/Mylip deletion on amyloid pathology and Aβ clearance. Idol is an E3 ubiquitin ligase which is a Liver X Receptors (LXRs) target and a post-transcriptional regulator of the low-density lipoprotein receptor (LDLR) in the periphery (Hong et al., 2014). Previous studies by David Holtzman’s group showed that the overexpression of LDLR in AD mice decreased amyloid deposition and increased Aβ clearance (Kim et al., 2009; Castellano et al., 2012). In the present article, the Tontonoz and Kim groups crossed Idol knockout mice to an AD model (APP/PS1) to examine amyloid pathology and Aβ clearance by microglia. The authors demonstrate that as in the periphery, the deletion of Idol increases brain LDLR protein level. They also show that Idol knockout mice have less soluble and insoluble amyloid, and decreased ApoE levels, and neuroinflammation. In a series of in vitro experiments using microglia from wild-type and Idol knockout mice, the authors prove that IDOL modulates cellular Aβ and ApoE uptake by microglia. Their conclusion is that DIOL is “a gatekeeper of LDLR-dependent ApoE and Aβ clearance in the brain.”
This is an interesting study that brings attention to a potentially new target for AD. It also raises a few questions: How does IDOL affect Aβ clearance via BBB? Also, how to reconcile the positive effect of LXR ligands on Aβ clearance through BBB (Fitz et al., 2010; Fitz et al., 2014) and by microglia (Jiang et al., 2008; Terwel et al., 2011) with the increased expression of LXR target, Idol?
References:
Castellano JM, Deane R, Gottesdiener AJ, Verghese PB, Stewart FR, West T, Paoletti AC, Kasper TR, DeMattos RB, Zlokovic BV, Holtzman DM. Low-density lipoprotein receptor overexpression enhances the rate of brain-to-blood Aβ clearance in a mouse model of β-amyloidosis. Proc Natl Acad Sci U S A. 2012 Sep 18;109(38):15502-7. Epub 2012 Aug 27 PubMed.
Fitz NF, Castranio EL, Carter AY, Kodali R, Lefterov I, Koldamova R. Improvement of memory deficits and amyloid-β clearance in aged APP23 mice treated with a combination of anti-amyloid-β antibody and LXR agonist. J Alzheimers Dis. 2014;41(2):535-49. PubMed.
Fitz NF, Cronican A, Pham T, Fogg A, Fauq AH, Chapman R, Lefterov I, Koldamova R. Liver X receptor agonist treatment ameliorates amyloid pathology and memory deficits caused by high-fat diet in APP23 mice. J Neurosci. 2010 May 19;30(20):6862-72. PubMed.
Hong C, Marshall SM, McDaniel AL, Graham M, Layne JD, Cai L, Scotti E, Boyadjian R, Kim J, Chamberlain BT, Tangirala RK, Jung ME, Fong L, Lee R, Young SG, Temel RE, Tontonoz P. The LXR-Idol axis differentially regulates plasma LDL levels in primates and mice. Cell Metab. 2014 Nov 4;20(5):910-8. PubMed.
Jiang Q, Lee CY, Mandrekar S, Wilkinson B, Cramer P, Zelcer N, Mann K, Lamb B, Willson TM, Collins JL, Richardson JC, Smith JD, Comery TA, Riddell D, Holtzman DM, Tontonoz P, Landreth GE. ApoE promotes the proteolytic degradation of Abeta. Neuron. 2008 Jun 12;58(5):681-93. PubMed.
Kim J, Castellano JM, Jiang H, Basak JM, Parsadanian M, Pham V, Mason SM, Paul SM, Holtzman DM. Overexpression of low-density lipoprotein receptor in the brain markedly inhibits amyloid deposition and increases extracellular A beta clearance. Neuron. 2009 Dec 10;64(5):632-44. PubMed.
University of British Columbia
ApoE has an undeniably important role in Alzheimer Disease (AD) pathogenesis and is best known for its role in Aβ metabolism. ApoE is the major lipoprotein expressed in the brain (Michaelson 2014). In its lifecycle as a lipoprotein, apoE is secreted by astrocytes and microglia, lipidated by the cholesterol and phospholipid transporter ABCA1, and lipid-laden apoE is taken up into target cells primarily by the low-density lipoprotein receptor (LDLR) (Huang and Mahley, 2014). ApoE levels and apoE lipidation status have pronounced effects on Aβ clearance. Genetic reduction of apoE levels reduces Aβ accumulation in a gene dose-dependent manner (Bales et al., 2002; Kim et al., 2011), and driving apoE uptake by increasing its lipidation (Koldamova et al., 2014) or LDLR overexpression (Castellano et al., 2012; Kim et al., 2009) also reduces Aβ accumulation. Whether these effects are determined by apoE-Aβ interactions (Namba et al., 1991) or competition of apoE with Aβ for clearance pathways (Verghese et al., 2013) is not yet fully understood.
Peter Tontonoz’s group has now identified an important role for Inducible Degrader of LDLR (IDOL) in the regulation of murine LDL, apoE, and Aβ metabolism. IDOL is an LXR-regulated E3 ubiquitin ligase that targets LDLR for degradation (Zhang et al., 2012). Choi et al. show that IDOL is widely expressed in murine microglia and neurons but not in astrocytes. Genetic ablation of IDOL has a gene-dose dependent increase in brain LDLR protein levels. In the APP/PS1 model of amyloidosis, IDOL deficiency reduces soluble and insoluble Aβ pools, lowers Aβ and amyloid deposits, reduces apoE protein levels, and attenuates microglial activation. In vitro, silencing of IDOL enhances Aβ uptake in N2a neuronal cells, apoE3-immortalized astrocytes, and BV2 microglial cells. IDOL-deficient primary microglia also exhibit elevated uptake of Aβ and reconstituted apoE lipoprotein particles.
These observations provide strong support for murine IDOL as a potent modulator of apoE and Aβ metabolism and of the brain’s innate neuroinflammatory pathways. Importantly, the results are consistent with a model where lowering apoE expression (via genetic ablation) or promoting apoE uptake (via increasing ABCA1 or LDLR levels) increases net Aβ clearance.
Although there is no question that lowering apoE levels reduces amyloid and Aβ burden in mice, whether direct reduction of apoE may compromise other cellular functions is now increasingly important to address. For example, LDLR-mediated uptake of apoE is important for neurite outgrowth (Pitas et al., 1998), and neuronal IDOL expression has recently been shown to be downregulated by pro-nerve growth factor (Do et al., 2015). Although apoE and IDOL deficiencies have similar effects on Aβ load, direct reduction of net apoE levels may compromise neurite outgrowth, whereas IDOL inhibition may preserve this. A closer look at neuronal IDOL function, especially with respect to synaptic and behavioral effects under baseline and neuronal stress conditions, are eagerly awaited.
Another salient point raised by Choi et al. relates to species-specific effects. In mice, IDOL deficiency affects LDLR levels in brain and adipose tissue but not in liver. Further, murine hepatic IDOL is unresponsive to LXR agonist treatment, whereas LXR activation induces IDOL expression in both human HepG2 cells and in primates, leading to altered plasma lipids and hepatic lipid homeostasis (Hong et al., 2014). Whether the ability of human HepG2 cells to uncouple lipoprotein uptake from cholesterol biosynthesis (Loregger et al., 2015) is also preserved in mice is a consideration for the translational potential of IDOL as a therapeutic target.
IDOL’s ability to regulate LDLR, apoE, and Aβ metabolism in mice defines a new pathway to explore IDOL inhibition as a potential AD therapeutic strategy. Additionally, understanding more about the species-specific functions of IDOL could shed considerable light on some of the challenges associated with translating results from murine models into humans, given their differences in lipid and immune regulation.
References:
Michaelson DM. APOE ε4: the most prevalent yet understudied risk factor for Alzheimer's disease. Alzheimers Dement. 2014 Nov;10(6):861-8. Epub 2014 Sep 10 PubMed.
Huang Y, Mahley RW. Apolipoprotein E: structure and function in lipid metabolism, neurobiology, and Alzheimer's diseases. Neurobiol Dis. 2014 Dec;72 Pt A:3-12. Epub 2014 Aug 27 PubMed.
Bales KR, Dodart JC, Demattos RB, Holtzman DM, Paul SM. Apolipoprotein E, amyloid, and Alzheimer disease. Mol Interv. 2002 Oct;2(6):363-75, 339. PubMed.
Kim J, Jiang H, Park S, Eltorai AE, Stewart FR, Yoon H, Basak JM, Finn MB, Holtzman DM. Haploinsufficiency of human APOE reduces amyloid deposition in a mouse model of amyloid-β amyloidosis. J Neurosci. 2011 Dec 7;31(49):18007-12. PubMed.
Koldamova R, Fitz NF, Lefterov I. ATP-binding cassette transporter A1: From metabolism to neurodegeneration. Neurobiol Dis. 2014 May 17; PubMed.
Castellano JM, Deane R, Gottesdiener AJ, Verghese PB, Stewart FR, West T, Paoletti AC, Kasper TR, DeMattos RB, Zlokovic BV, Holtzman DM. Low-density lipoprotein receptor overexpression enhances the rate of brain-to-blood Aβ clearance in a mouse model of β-amyloidosis. Proc Natl Acad Sci U S A. 2012 Sep 18;109(38):15502-7. Epub 2012 Aug 27 PubMed.
Kim J, Castellano JM, Jiang H, Basak JM, Parsadanian M, Pham V, Mason SM, Paul SM, Holtzman DM. Overexpression of low-density lipoprotein receptor in the brain markedly inhibits amyloid deposition and increases extracellular A beta clearance. Neuron. 2009 Dec 10;64(5):632-44. PubMed.
Namba Y, Tomonaga M, Kawasaki H, Otomo E, Ikeda K. Apolipoprotein E immunoreactivity in cerebral amyloid deposits and neurofibrillary tangles in Alzheimer's disease and kuru plaque amyloid in Creutzfeldt-Jakob disease. Brain Res. 1991 Feb 8;541(1):163-6. PubMed.
Verghese PB, Castellano JM, Garai K, Wang Y, Jiang H, Shah A, Bu G, Frieden C, Holtzman DM. ApoE influences amyloid-β (Aβ) clearance despite minimal apoE/Aβ association in physiological conditions. Proc Natl Acad Sci U S A. 2013 May 7;110(19):E1807-16. PubMed.
Zhang L, Reue K, Fong LG, Young SG, Tontonoz P. Feedback regulation of cholesterol uptake by the LXR-IDOL-LDLR axis. Arterioscler Thromb Vasc Biol. 2012 Nov;32(11):2541-6. Epub 2012 Aug 30 PubMed.
Pitas RE, Ji ZS, Weisgraber KH, Mahley RW. Role of apolipoprotein E in modulating neurite outgrowth: potential effect of intracellular apolipoprotein E. Biochem Soc Trans. 1998 May;26(2):257-62. PubMed.
Do HT, Bruelle C, Pham DD, Jauhiainen M, Eriksson O, Korhonen LT, Lindholm D. Nerve growth factor (NGF) and pro-NGF increase low-density lipoprotein (LDL) receptors in neuronal cells partly by different mechanisms: role of LDL in neurite outgrowth. J Neurochem. 2015 Oct 20; PubMed.
Hong C, Marshall SM, McDaniel AL, Graham M, Layne JD, Cai L, Scotti E, Boyadjian R, Kim J, Chamberlain BT, Tangirala RK, Jung ME, Fong L, Lee R, Young SG, Temel RE, Tontonoz P. The LXR-Idol axis differentially regulates plasma LDL levels in primates and mice. Cell Metab. 2014 Nov 4;20(5):910-8. PubMed.
Loregger A, Cook EC, Nelson JK, Moeton M, Sharpe LJ, Engberg S, Karimova M, Lambert G, Brown AJ, Zelcer N. A MARCH6 and IDOL E3 ubiquitin ligase circuit uncouples cholesterol synthesis from lipoprotein uptake in hepatocytes. Mol Cell Biol. 2015 Nov 2; PubMed.
Washington University
There is strong evidence that the LDL receptor (LDLR) regulates apoE levels in the brain. Increasing LDLR decreases apoE and Aβ and decreasing LDLR has the opposite effect. These effects may occur via apoE’s ability to modulate Aβ levels, or potentially, via direct effects of LDLR, Aβ clearance. Thus, increasing LDLR may be a good therapeutic target in AD. In this paper from the Kim and Tontonoz labs, they study a regulator of cellular levels of LDLR known as IDOL. They clearly demonstrate the IDOL regulates LDLR levels in the brain. IDOL knockout mice have increased LDLR levels in the brain and when crossed to APP transgenic mice, there are significantly lower levels of apoE, insoluble Aβ, and microgliosis. The results appear strong, solid, and clear. If IDOL can be targeted in the brain, this seems like a strong candidate for a target for AD therapeutics. This work also strengthens the idea that increasing LDLR in the brain is an AD therapeutic target. Understanding the details of how LDLR decreases Aβ levels still needs to be sorted out.
Make a Comment
To make a comment you must login or register.