Past Webinar
Adding Dynamics and Nuance to Alzheimer’s Staging
Quick Links
Introduction
Being able to image amyloid, glucose metabolism, atrophy, even network connectivity in living patients has been a boon for those trying to stage the progression of Alzheimer’s disease. Using magnetic resonance imaging (MRI) and positron emission tomography (PET) with different tracers, many labs have shown that, as the disease progresses, amyloid increases, metabolism decreases, networks disconnect, and the brain shrinks.
But these changes don’t happen in the same sequence throughout the brain. When scientists use several imaging modalities together in the same patient and look at various regions of the brain, they see different alterations emerge at distinct stages of disease, depending on the brain region observed. What goes up in one region goes down in another, yielding a more complex picture of disease progression than previously envisioned. How are all these changes related to one another? Will putting disparate snapshots of the brain together afford us a more accurate view of what actually happens in the disease? With all that imaging going in the field, is a movie of what changes where and when within reach?
On 5 January 2012, Alzforum held a Webinar discussion on the topic. Gael Chételat was joined by Anna Caroli, Keith Johnson, Beth Mormino, Prashanthi Vemuri, and other imaging experts to discuss advances in multimodal neuroimaging of Alzheimer’s disease.
- Gael Chételat's Presentation
- Anna Caroli's Presentation
- Keith Johnson's Presentation
- Beth Mormino's Presentation
- Prashanthi Vemuri's Presentation
Media
Background
Background Text
By Gael Chételat
Adding Nuance to Staging Diagrams: Integrated Brain Imaging Emphasizes Regional Differences in What Changes When on the Long Descent Into Alzheimer’s
Numerous neuroimaging techniques have been developed during the past 20 years, allowing scientists to unravel the spectrum of brain alterations that occurs in Alzheimer’s disease (AD). These different measures, which include magnetic resonance imaging (MRI), positron emission tomography (PET) with fluorodeoxyglucose (FDG), and others, are complementary, as they probably reflect different underlying pathological processes. Most of these changes have been independently and thoroughly investigated in AD, but only recently have scientists begun to look at the relationships among them to try to understand how changes over time in different brain regions might eventually fit together into a more complete picture of brain injury in preclinical AD. Multimodal and longitudinal imaging studies of different brain regions are a way forward here. Such work, undertaken by our group and others, is beginning to reveal a more complex picture of the evolution of AD pathology than articulated in the initial proposed staging diagrams of biomarker change in AD. A more nuanced model is starting to emerge whereby AD involves a variety of pathogenic and compensatory mechanisms that change according to brain region and stage of disease.
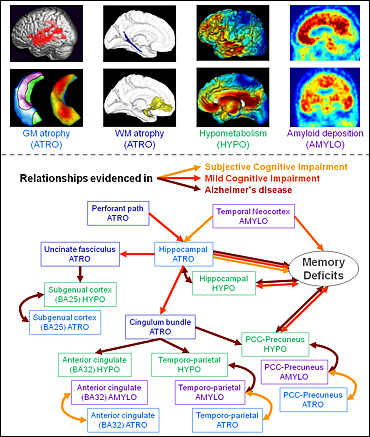
Different neuroimaging techniques reveal the spectrum of changes that occurs in the Alzheimer’s disease brain. The relationships among these changes differ depending on brain region and stage of disease. Image credit: Gael Chételat
Several MRI studies have highlighted a predominance of hippocampal region atrophy early in the presymptomatic stage of AD. These studies showed that atrophy progresses to the temporal cortex, temporoparietal and posterior cingulate cortex (PCC), and then to the frontal cortex as the disease evolves. On the other hand, studies using resting-state FDG-PET, which measures change in glucose metabolism and is an indirect measure of neuronal dysfunction (see ARF related news story), have shown that hypometabolism predominates in the PCC-precuneus region and temporoparietal area before extending to medial orbitofrontal areas of the brain; the earliest changes are again observed at the presymptomatic stage.
More recently, there has been an explosion of interest in the in-vivo assessment of amyloid deposition in the brain, thanks to the development of radiotracers such as the Pittsburgh Compound B (PIB), a fluorescent analog of thioflavin T that binds amyloid-β (Aβ) in the brain, or 18F-AV-45 (florbetapir F-18), an agent currently under consideration by the U.S. Food and Drug Administration (FDA). Studies have shown a widespread pattern of amyloid deposition, especially in medial frontal and parietal areas of the brain, but also extending to almost the whole neocortex. This pattern occurs in about one-third of cognitively normal people, a finding that is often interpreted as a sign of presymptomatic AD. Thus, amyloid deposition, brain atrophy, or hypometabolism changes were shown to appear before any measurable clinical decline.
Now, the AD field is rapidly moving to multimodal neuroimaging studies, whereby several biomarkers are measured simultaneously in the same patient, allowing scientists to assess relationships among markers, as well as how these markers change over time and in what sequence. Several dynamic models have been proposed to illustrate the chronology of different AD biomarker changes (i.e., their evolution over the course of the disease) (Jack et al., 2010; Weiner et al., 2010; Frisoni et al., 2010; Petersen, 2010; Perrin et al., 2009). These models differ in several aspects, such as the shapes of the curves, the time when various changes start, and if and when they plateau, but there are also clear similarities. Almost all models propose the same chronology, with amyloid deposition coming first, followed by neuronal dysfunction, then atrophy, and then cognitive decline. Importantly, according to the models proposed thus far, this chronology does not change according to brain regions.
The amyloid hypothesis that predominates current research has significantly inspired these models. They typically predict that Aβ occurs first in the neocortex prior to any other changes. Indeed, there is little doubt that in the neocortex, Aβ appears before neurofibrillary tangles (NFTs). However, things become more complicated if we consider separately different regions of the brain. For instance, the stages of AD evolution as per Braak and Braak predict that in the hippocampus, NFTs occur before Aβ deposition (Braak and Braak, 1991; see also online seminar).
It thus appears that the chronology of disease-associated events is region specific. Most AD biomarkers are multi-determined. In other words, they are induced by several underlying mechanisms that operate to different degrees in various areas of the brain, which may reflect different vulnerability to disease. We therefore argue that distinct biomarkers should be considered separately in different brain regions in order to disentangle the chronology of pathogenic events in AD and to help explain the dynamic changes and relationships among markers.
For instance, we pointed out the discordance between the pattern of brain atrophy and that of hypometabolism. Specifically, we observed early and marked atrophy in the hippocampus, in contrast to a relative metabolic preservation of this brain region; we found the opposite was true in the PCC (Chételat et al., 2008; see also ARF related news story). This observation led us to propose that different underlying pathological processes occur in the hippocampus and the PCC, with hippocampal atrophy probably related to the early appearance of NFTs in this region, while PCC hypometabolism would reflect disruption of hippocampal-PCC connectivity. This so-called “disconnection hypothesis” received support from cross-sectional studies showing a relationship among hippocampal atrophy, white matter tract alteration, and hypometabolism in the posterior associative neocortical areas (Villain et al., 2008). In a subsequent longitudinal study, we showed that there is a causal relationship among these three pathological events. Specifically, hippocampal atrophy progressively leads to disruption of the cingulum bundle and uncinate fasciculus, which in turn leads to glucose hypometabolism of the cingulate and subgenual cortices (Villain et al., 2010).
This contrast between hypometabolism-predominant areas versus atrophy-predominant areas has been further strengthened by the addition of a third modality, that is, amyloid deposition. In a recent study, we clustered brain regions according to their relative degree of atrophy, hypometabolism, and amyloid deposition. Based on these markers, we distinguished three sets of brain regions: 1) the medial temporal regions, where atrophy exceeded hypometabolism and Aβ load was low; 2) the temporoparietal cortex, precuneus, and posterior cingulate cortex, where all three alterations were marked, but hypometabolism and Aβ load exceeded atrophy; and 3) the frontal regions, where Aβ deposition clearly predominated, while structural and metabolic alterations were minimal. Again, these findings suggest that different underlying pathogenic mechanisms occur in these three sets of brain regions. This would then account for their differential sensitivity to atrophy, hypometabolism, and amyloid deposition.
To further understand these underlying mechanisms, we assessed the local relationships among the three modalities by measuring the region-to-region or voxel-to-voxel correlations between pairs of measurements in patients with probable AD. We found relatively marked and widespread correlations between atrophy and hypometabolism in the hippocampal region, temporal and temporoparietal cortex, posterior cingulated-precuneus, and frontal cortex. This suggests that atrophy and hypometabolism are related and co-varied in these regions; however, additional mechanisms may amplify or attenuate either atrophy or hypometabolism in specific brain areas. These may include disconnection processes in regions that show more hypometabolism than atrophy. On the other hand, in medial temporal regions, the predominance of atrophy may be related to the presence of NFTs and potential functional compensatory processes the brain uses in the earliest stages of disease.
We found that correlations between hypometabolism and amyloid deposition were limited. They only occurred in the PCC-precuneus region and part of the temporoparietal cortex. These two modalities showed no relationship in the frontal cortex, a region where amyloid deposition is particularly marked. One previous study even showed a negative correlation between amyloid deposition and frontal metabolism in patients with mild cognitive impairment (MCI); in these patients, greater amyloid deposition was related to higher metabolism in the frontal cortex (see ARF related news story on Cohen et al., 2009). It is possible that the frontal cortex employs functional compensatory mechanisms (probably at later stages of disease than the compensatory mechanisms in the hippocampus) to maintain or increase glucose metabolism despite amyloid deposition. This hypothesis would fit with data from functional MRI (fMRI) studies showing increased brain activity in the frontal cortex in MCI or AD patients compared to controls (Schwindt and Black, 2009). Consistent with these results, one previous study in our lab showed that a decrease in metabolism over time in the ventromedial orbitofrontal cortex of MCI patients progressing to AD correlated to increased metabolism in the dorsomedial frontal cortex (Fouquet et al., 2009).
We find no significant voxel-to-voxel correlations between atrophy and amyloid deposition in patients with probable AD. This conclusion was consistent with findings obtained from the Australian Imaging, Biomarkers and Lifestyle (AIBL; Ellis et al., 2009) cohort, where we showed that there was no relationship between amyloid deposition and atrophy at either the AD or MCI stages (Chételat et al., 2010). However, we found significant relationships between these two biomarkers in the anterior and posterior cingulate and temporoparietal cortex at even earlier stages of disease (i.e., in patients with subjective memory complaints). Altogether, these findings suggest that amyloid and atrophy are related only in earliest, presymptomatic stages of disease and only in regions of highest amyloid deposition (i.e., the anterior cingulate cortex, PCC, temporoparietal cortex).
As hippocampal atrophy was not linked to local amyloid deposition at any stage of disease, we wondered whether it might be linked to amyloid deposition elsewhere in the brain. Indeed, when we assessed the correlation between hippocampal atrophy and amyloid deposition throughout the brain, both voxelwise and using a region-of-interest approach, we consistently found a specific relationship between hippocampal atrophy and amyloid deposition in the temporal neocortex in asymptomatic people.
To sum up these findings, we found three sets of brain regions that are quite different in their relative degree of atrophy, hypometabolism, and amyloid deposition. In the temporal, temporoparietal, and PCC-precuneus regions, hypometabolism and amyloid deposition exceeded atrophy; in these regions, the three modalities were highly correlated, and the higher degree of hypometabolism compared to atrophy could be due to amyloid deposition and the disruption of PCC connectivity to the hippocampus. In the hippocampus, atrophy predominated; atrophy and hypometabolism were related, but the higher amount of atrophy may be due to the presence of NFTs, distant (temporal neocortical) amyloid deposition, and local functional compensation. Finally, in the frontal cortex, atrophy was related to local hypometabolism and amyloid deposition, and part of the frontal cortex was disconnected from the hippocampus. The multiple inputs to this region and its possible lower sensitivity to amyloid deposition, together with functional compensatory processes, may explain the profile of atrophy, hypometabolism, and amyloid deposition.
In conclusion, we argue for a multipronged cause of AD-related brain neural injury. The differential expression of several underlying mechanisms across brain regions determines the relative degree of resulting atrophy, hypometabolism, and amyloid deposition. We think that there is not a single cause and sequence of events in AD throughout the brain, but rather that both causes and chronology are region specific.
References
News Citations
- Add to ADNI: Imaging Amyloid and Analyzing the Genome
- Paris: Beyond Genomics, French Science Draws on Populations, Patients
- Research Brief: Compensation or Constitution—Metabolism in MCI
Paper Citations
- Jack CR, Knopman DS, Jagust WJ, Shaw LM, Aisen PS, Weiner MW, Petersen RC, Trojanowski JQ. Hypothetical model of dynamic biomarkers of the Alzheimer's pathological cascade. Lancet Neurol. 2010 Jan;9(1):119-28. PubMed.
- Weiner MW, Aisen PS, Jack CR, Jagust WJ, Trojanowski JQ, Shaw L, Saykin AJ, Morris JC, Cairns N, Beckett LA, Toga A, Green R, Walter S, Soares H, Snyder P, Siemers E, Potter W, Cole PE, Schmidt M, . The Alzheimer's disease neuroimaging initiative: progress report and future plans. Alzheimers Dement. 2010 May;6(3):202-11.e7. PubMed.
- Frisoni GB, Fox NC, Jack CR, Scheltens P, Thompson PM. The clinical use of structural MRI in Alzheimer disease. Nat Rev Neurol. 2010 Feb;6(2):67-77. PubMed.
- Petersen RC. Alzheimer's disease: progress in prediction. Lancet Neurol. 2010 Jan;9(1):4-5. PubMed.
- Perrin RJ, Fagan AM, Holtzman DM. Multimodal techniques for diagnosis and prognosis of Alzheimer's disease. Nature. 2009 Oct 15;461(7266):916-22. PubMed.
- Chételat G, Desgranges B, Landeau B, Mézenge F, Poline JB, De la Sayette V, Viader F, Eustache F, Baron JC. Direct voxel-based comparison between grey matter hypometabolism and atrophy in Alzheimer's disease. Brain. 2008 Jan;131(Pt 1):60-71. PubMed.
- Villain N, Desgranges B, Viader F, De la Sayette V, Mézenge F, Landeau B, Baron JC, Eustache F, Chételat G. Relationships between hippocampal atrophy, white matter disruption, and gray matter hypometabolism in Alzheimer's disease. J Neurosci. 2008 Jun 11;28(24):6174-81. PubMed.
- Villain N, Fouquet M, Baron JC, Mézenge F, Landeau B, De la Sayette V, Viader F, Eustache F, Desgranges B, Chételat G. Sequential relationships between grey matter and white matter atrophy and brain metabolic abnormalities in early Alzheimer's disease. Brain. 2010 Nov;133(11):3301-14. PubMed.
- Cohen AD, Price JC, Weissfeld LA, James J, Rosario BL, Bi W, Nebes RD, Saxton JA, Snitz BE, Aizenstein HA, Wolk DA, Dekosky ST, Mathis CA, Klunk WE. Basal cerebral metabolism may modulate the cognitive effects of Abeta in mild cognitive impairment: an example of brain reserve. J Neurosci. 2009 Nov 25;29(47):14770-8. PubMed.
- Schwindt GC, Black SE. Functional imaging studies of episodic memory in Alzheimer's disease: a quantitative meta-analysis. Neuroimage. 2009 Mar 1;45(1):181-90. PubMed.
- Fouquet M, Desgranges B, Landeau B, Duchesnay E, Mézenge F, De la Sayette V, Viader F, Baron JC, Eustache F, Chételat G. Longitudinal brain metabolic changes from amnestic mild cognitive impairment to Alzheimer's disease. Brain. 2009 Aug;132(Pt 8):2058-67. PubMed.
- Ellis KA, Bush AI, Darby D, De Fazio D, Foster J, Hudson P, Lautenschlager NT, Lenzo N, Martins RN, Maruff P, Masters C, Milner A, Pike K, Rowe C, Savage G, Szoeke C, Taddei K, Villemagne V, Woodward M, Ames D, . The Australian Imaging, Biomarkers and Lifestyle (AIBL) study of aging: methodology and baseline characteristics of 1112 individuals recruited for a longitudinal study of Alzheimer's disease. Int Psychogeriatr. 2009 Aug;21(4):672-87. PubMed.
- Chételat G, Villemagne VL, Bourgeat P, Pike KE, Jones G, Ames D, Ellis KA, Szoeke C, Martins RN, O'Keefe GJ, Salvado O, Masters CL, Rowe CC, . Relationship between atrophy and beta-amyloid deposition in Alzheimer disease. Ann Neurol. 2010 Mar;67(3):317-24. PubMed.
Other Citations
External Citations
Further Reading
Papers
- Bookheimer SY, Strojwas MH, Cohen MS, Saunders AM, Pericak-Vance MA, Mazziotta JC, Small GW. Patterns of brain activation in people at risk for Alzheimer's disease. N Engl J Med. 2000 Aug 17;343(7):450-6. PubMed.
- Skoog I. Detection of preclinical Alzheimer's disease. N Engl J Med. 2000 Aug 17;343(7):502-3. PubMed.
Panelists
-
 Gael Chetelat
Cyceron
Gael Chetelat
Cyceron
-
 Anna Caroli, PhD (neuroscience)
IRCCS Centro san Giovanni di Dio - Fatebenefratelli
Anna Caroli, PhD (neuroscience)
IRCCS Centro san Giovanni di Dio - Fatebenefratelli
-
 Keith Johnson, M.D.
MGH, BWH, HMS
Keith Johnson, M.D.
MGH, BWH, HMS
-
 Elizabeth Mormino, Ph.D.
Stanford University School of Medicine
Elizabeth Mormino, Ph.D.
Stanford University School of Medicine
-
 Prashanthi Vemuri, Ph.D.
Mayo Clinic and Foundation
Prashanthi Vemuri, Ph.D.
Mayo Clinic and Foundation
Comments
No Available Comments
Make a Comment
To make a comment you must login or register.