Past Webinar
Neuroinflammation—A Prelude to Alzheimer's?
Quick Links
Introduction
Neuroinflammation plays a major role in Alzheimer's disease, but whether it represents a cause or a response to pathology is unclear. In the November 27 Nature Reviews Neurology, Dimitrije Krstic and Irene Knuesel argue that age-related chronic inflammation occurs early in the disease and puts neurons under undue stress. The cells respond by accumulating misfolded tau, which in turn derails axonal transport, leading to further neuronal damage, including accumulation of Aβ. They suggest that focusing on this "inflammation hypothesis of AD" could herald new directions in AD research and therapy.
In this Webinar, Knuesel, from the University of Zurich, Switzerland, explained her concepts. John Breitner, McGill University; Michael Heneka, University of Bonn; Frank Heppner, Charité Universitaetsmedizin Berlin; and Terrence Town, Cedars-Sinai Medical Center, Los Angeles, joined Knuesel for a panel discussion.
We thank Nature Reviews Neurology for giving Alzforum readers temporary free access to this paper:
Krstic D, Knuesel I. Deciphering the mechanism underlying late-onset Alzheimer disease. Nat Rev Neurol. 2012 Nov 27.
- Listen to the full Webinar
- Irene Knuesel's Presentation
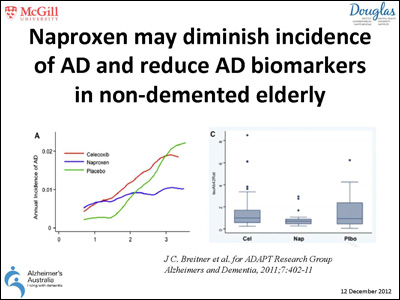
- John Breitner's Slide
- Michael Heneka's Presentation
- Frank Heppner’s Presentation
- Terrence Town's Presentation
Background
Background Text
by Tom Fagan
Scientists have long recognized the potential of slowing or halting Alzheimer's disease by suppressing the immune system and preventing neuroinflammation. Though non-steroidal anti-inflammatory drugs (NSAIDs) did not seem to help those with AD (see ARF related news story), and the ADAPT prevention trial of NSAIDs was halted early due to concerns about adverse side effects, substantial epidemiological evidence suggests that anti-inflammatory agents protect against the disease (see ARF related news story). John Breitner's re-analysis of the ADAPT trial data also hints that protection is afforded to patients taking NSAIDs after some time (see ARF related news story and Breitner et al., 2011). Some researchers in the field have not given up hope that reining in inflammation may successfully prevent or at least slow down AD pathology.
Against this backdrop, geneticists and cellular and molecular biologists have begun to flesh out how the immune system dovetails with AD pathology. Genomewide association studies identified complement receptor 1, part of the innate immune system, as a risk factor for sporadic AD (see ARF related news story), and just last month rare genetic variants in the gene for the microglial receptor TREM2 emerged as strong risk factors for late-onset disease as well (see ARF related news story). Working with mouse models, others, including Terrence Town, showed that blocking inflammatory responses in the innate immune system attenuates AD-like pathology (see ARF related news story). More recently, Frank Heppner and colleagues reported that antibodies to inflammatory interleukins 12 and 23 also prevent plaque buildup in mouse models (see ARF related news story). The latest work from Michael Heneka's lab suggests using PPARγ agonists to tone down inflammation while boosting microglial uptake of Aβ, as a potential therapeutic approach (see ARF related news story).
With evidence building for neuroinflammatory signals being key players in pathology, the question of cause and effect takes on greater importance. Does neuroinflammation drive, or is it driven by, other pathologies, including senile plaques and neurofibrillary tangles? Irene Knuesel has examined this using mouse models. She discovered that a neuroinflammatory stimulus given to young triple transgenic AD mice dramatically boosted plaque production 11 months later. Intriguingly, she also found that an immune stimulus given to wild-type mice early in life increases their susceptibility to AD-like pathology when given a second immune stimulus as adults (see ARF related news story). The findings suggest that neuroinflammation lays down the conditions for AD pathology to flourish, and that factors that fan the immune flames, including infection, disease, or even age, may be major risk factors for disease.
Q: Why did you look during gestation day 17 (GD17), and why did you use polyI:C?
Irene Knuesel: The GD17 period of embryonic development is characterized by the onset of synaptogenesis as shown morphologically, as well as by the induction of several genes involved in synaptogenesis. Our previous studies confirmed that disturbances during this crucial developmental phase result in long-term neurochemical and behavioral changes in the offspring: Offspring of GD17 immune-challenged mouse dams show cognitive impairments and reduction in neurogenesis in adulthood as compared to those challenged with saline controls (Meyer et al., 2006; Meyer et al., 2008). We therefore reasoned that these mice could be ideal to test the concept of “cognitive reserve,” i.e., the hypothesis that neuronal networks that are less efficient or have lower capacity may be more vulnerable to disruption, for example, induced by chronic inflammatory conditions.
GD17 is also a time window when microglia populate the embryonic brain, i.e., the initiation of embryonic innate immunity. In contrast to GD9, late-gestational embryos also contribute to the cytokine response triggered by polyI:C (Meyer et al., 2006). Work from Paul Patterson’s lab also confirmed that the GD9-treated offspring—in contrast to the GD17—do not show long-term changes in several cytokines and chemokines in adulthood.
PolyI:C, a synthetic, double-stranded RNA (mimicking RNA viruses) has been widely used to induce Toll-like receptor 3-mediated cytokine/chemokine expression in both in-vitro and in-vivo applications. It is conceivable that a bacterial-like infection has similar long-lasting effects. However, we have not tested this in our mice.
Q: Does a second polyI:C stimulus cause cognitive impairments in APP mice?
Irene Knuesel: We did not apply multiple polyI:C stimuli so far to the TgAD mice, and therefore cannot answer this highly relevant question.
We applied a prenatal infection to our TgAD mice (ArcAβ and 3xTgAD lines). This manipulation resulted in a mild acceleration of the plaque pathology; however, genetic factors (i.e., promoters) appeared to be the more prominent driver for the plaque initiation. Based on these findings, we applied adult systemic infections to the TgAD mice to test the hypothesis that the progression of the pathology may be affected by the immune challenge. And as highlighted in our paper (Krstic et al., 2012), it indeed does! So it appears as if the immune challenge during adulthood/aging aggravates the neuropathology in predisposed mice (i.e., wild-type mice exposed to a prenatal immune challenge and Tg mice harboring a genetic “predisposition”).
Q: How does CD45 microglial reactivity or Iba/1 microglial signal change after a second polyI:C stimulus in APP mice?
Irene Knuesel: See my answer above: So far, we did not apply multiple immune challenges in TgAD mice. I expect also an aggravation of both the neuropathology and the cognitive performance. We should definitively test this experimentally.
Q: What makes this proposed mechanism specific to Alzheimer's disease?
Irene Knuesel: Considering the overlap between neuropathological hallmarks and the development of dementia in late-onset neurodegenerative diseases, it is tempting to speculate that the hypothetical sequence of events proposed in our review is not specific to late-onset Alzheimer’s disease, but relevant for other aging-associated forms of neurodegenerative diseases as well, such as Parkinson’s disease or frontotemporal dementia. It is conceivable that the initiating event in elderly individuals with their unique life history is completely stochastic. Based on the selective connectivity of the affected groups of neurons, the progression of the disease (and accumulation of distinct aggregation-prone proteins), and hence the behavioral symptoms, might then follow a different path and culminate in an AD, PD, or FTD phenotype.
Q: A provocative question to all. To date, we have been able to cure "AD" more than 300 times in mice, but zero times in humans. In light of data from Streit (Streit et al., 2009) showing that microglia in AD patients appear dystrophic rather than activated, do you think that the data on microglia in AD transgenic mice, which look healthy and activated, translate into patients?
Terrence Town: I agree that it has been difficult to translate results from transgenic mouse models into therapies for human AD. In my mind, at least, this is why we need much better, and more stringent, AD animal models. Developing such models may necessitate changing the species, and my group is currently working on a rat model that seems to recapitulate most—if not all—of the pathophysiologic hallmarks of the disease. Regarding the comment about microglia, this is a somewhat controversial area. In humans, some (like Streit) maintain that microglia are senescent, while others have evidence for activation of these cells. I’m not sure I agree that the microglia in the transgenics are "healthy," though. By immune/inflammatory markers and morphology, these cells appear to be in a proinflammatory state.
Frank Heppner: This is an important and often overlooked point. Although microglia in AD transgenic mouse models don’t exhibit the same morphological characteristics described by Streit et al., this is likely because the lifespan of mice is too short for these changes to occur and become fully transparent. However, data published by the same group (Flanary et al. 2007) show that rodents do exhibit signs of cellular senescence in the form of age-related attrition of microglia telomeres, for example, and unpublished microarray studies by our group analyzing isolated microglia show that microglia in AD mouse models do exhibit accelerated age-related genetic changes. Furthermore, using in-vivo and ex-vivo analyses, we have tested microglia functions (phagocytosis, environmental surveillance, and response towards injury) and found that microglia functions progressively decline during the course of AD-like pathology, and that this decline relates to the extent of amyloid plaque burden (Krabbe et al., submitted for publication). This would suggest that, although microglia in transgenic mouse models are not fully "senescent," processes similar to those in the human condition are likely at play; i.e., microglia in AD mouse models display a phenotype best characterized as dysfunction/lack of function. So, while animal models cannot fully mimic the human condition, even in this aspect, there is likely much to learn from them, though admittedly, a great deal of work to do.
Q: I have not heard much about the blood-brain barrier (BBB) in neuroinflammation in AD, or about trauma as the most prominent environmental risk factor for AD. Can the speakers address?
Irene Knuesel: The experimental approaches with regard to the BBB in AD have mainly centered on the effect of Aβ on integrity and functions of the barrier. It would be very relevant to address how chronic inflammatory conditions and aging alter astrocytic functions at the BBB. Although it is well known that disruption of the BBB induces a strong neuroinflammatory reaction and oxidative stress, it is equally conceivable that the decline in glial support and protection during aging might be accelerated by chronic neuroinflammatory stimuli, and then that might trigger the breakdown of the BBB in advanced/late stages of AD.
Accumulating experimental evidence from clinical and preclinical studies suggests that in traumatic brain injury (TBI) models, there is a substantial increase in APP expression and accumulation at the sites of injury, accompanied by a local increase in Aβ production and amyloid plaque deposition (Stone et al., 2001; Smith et al., 2003). Interestingly, ApoE4 carriers have a significantly increased risk to develop dementia after head injury (e.g., Sundstrom et al., 2007), indicating that the recovery from neuronal damage is profoundly impaired in these individuals. These findings are in line with our model highlighting the putative difference between ApoE4 and ApoE3 carriers with respect to counteractive and protection mechanisms that prevent the spread of axonal leakage to neighboring neurons. Of interest are also the findings from animal models that show TBI-induced synaptic degeneration in APP knockout and impaired regeneration in ApoE4 knock-in mice, pointing to the fundamental role of ApoE and APP in stress-related neuronal repair and protection.
Q: What happens if you will be able to knock out the microglia?
Terrence Town: This question is probably best addressed by Frank Heppner, who has done this very experiment in transgenic mice. Much to our surprise, nothing happened in terms of amyloid pathology!
Frank Heppner: We have done this experiment (Grathwohl et al., 2009; and see ARF related news story), temporarily ablating microglia for up to 30 days. To our surprise, this manipulation—at least in APP/PS1 mice (Radde et al., 2006)—did not affect formation and maintenance of β amyloid plaques. Since our approach to deplete microglia in vivo does not allow for longer time periods of microglia ablation, we cannot test what happens when microglia are gone for a prolonged period of time (i.e., >30 days). Thus, one should (and must) not conclude from these experiments that microglia do not play a role in AD pathogenesis! In my opinion, it very much depends on how long microglia can (or cannot) act, and when they can (or cannot) act. Beyond that, we also must keep in mind that the picture may look different in other AD-like models.
Q: I'm concerned with the staging argument with respect to therapy. We need to keep in mind that this is a progressive disorder with different regions of the brain existing at different stages at a single point in time. The net effects of anti-inflammatory therapies may be helpful and harmful with regional specificity. Comments?
Terrence Town: I think this is an excellent point, and a very real possibility. We now know that microglia and macrophages have different ways of handling Aβ—for example, Gary Landreth and others have shown that fibrillar Aβ is cleared through receptor-mediated phagocytosis, while soluble forms of the peptide rely on fluid phase macropinocytosis for clearance. Further, we know that Aβ is a meta-stable peptide, existing in different forms simultaneously throughout the brain. Given this, it is entirely possible that a particular microglia-targeted therapy may be beneficial in one brain region, but have limited or even detrimental effect in another brain area. But, rather than a negative, I see your question as a positive—an opportunity for basic biologic investigation into mechanisms of microglia-targeted therapeutics.
Q: Thanks for this highly interesting Webinar. There are some studies indicating that mutated presenilins may have a direct effect on microglial reactivity capacities. Could you comment on whether PS/APP or 3xTg mice are good models for microglial analysis?
Terrence Town: That is correct—there is evidence that mutant presenilins impact immune system function and inflammation. In terms of the transgenics, though, to the extent that we seek to model familial AD, we are left to rely on the presenilins and APP. I think it’s a reasonable assumption that effects of mutant presenilin/APP on microglia in the animal models would approximate similar effects in brains of familial AD patients.
Frank Heppner: Depending on the promoter used, some mouse models are probably not good for the study of microglia function. For example, work by Sam Sisodia (Choi et al., 2008) showed that microglia in mice expressing human presenilin 1 variants linked to FAD under the prion promoter exhibited impaired neurogenesis, which turned out to be due to alterations in microglia function resulting from transgene expression in these cells. The neurogenesis phenomenon may or may not translate to humans. Thus, a careful selection of the transgenic model of choice is required. In these instances, I feel that we still can learn a lot from these models, despite the limitation that a model is just a model…. Moreover, Irene Knuesel’s gestation day 17 (GD17) polyI:C model (Krstic et al., 2012) may also add to the repertoire of available models for assessing microglia function in AD.
Q: This is quite interesting. What are the implications for therapy? One would think that the anti-amyloid antibodies would produce some level of inflammation and make patients worse. But, this doesn't seem to happen. The antibodies seem to do nothing. Similarly, COX2 inhibitors, which are anti-inflammatory, don't seem to offer any benefit.
Terrence Town: Actually, the Elan/Wyeth AN1792 immunotherapy trial was halted when ~6 percent of patients developed a severe form of brain inflammation known as aseptic meningoencephalitis—so, at least in a subset of patients, inflammation was provoked by the treatment. Despite this, some of the patients clearly showed evidence of plaque removal upon pathological investigation, as detailed by James Nicoll and colleagues.
Q: What is your view of the dual effect of inflammation: beneficial versus deleterious?
Terrence Town: This is a key question, and the answer can likely be summed up in three words: context, context, context…! What we are starting to see is that a number of factors conspire to determine whether a particular inflammatory stressor leads to a beneficial or detrimental response. For example, the timing, duration, and particular inflammatory cascade(s) activated all play into the type of inflammatory response. Embedded within your question is a call for deep, basic biological investigation into which pathways are beneficial, so that we may use this knowledge to inform microglial-directed therapeutic strategies in moving forward.
Irene Knuesel: Couldn’t agree more: We need more investigations into the role of microglia during physiological and pathophysiological forms of aging in order to understand their protective and potentially harmful effects on neurons.
Frank Heppner: First of all, it is important to note that inflammation already implies a rather adverse status of the immune system; we should rather use terms such as “modulation” or “skewing” of the immune system. On top of that, as pointed out on my final slide in the Webinar, I think it very much depends when certain immune molecules (including those which are microglia derived) act, and for how long they act in the course of the disease. In turn, it matters which specific immune components will be manipulated, when, and for how long. Thus, we need to dissect the impact of distinct immune components, molecules, and pathways in much more detail, in a fashion similar to what we have done most recently (Vom Berg et al., 2012 and see ARF related news story). Consequently, there is probably no such thing as exclusively “good“ or “bad“ microglia/state of the immune system/“inflammation” in AD.
Q: Does the presence of acetylcholine influence microglia cells in AD by suppression or activation?
Irene Knuesel: It is known that the α7-containing nicotinic acetylcholine receptors (α7nAChRs) exert anti-inflammatory properties via vagal nerve-mediated reduction of TNF-α release from peripheral macrophages (see Wang et al., 2003). Recent evidence suggests that nAChRs may also exert neuroprotection in mouse models of Parkinson’s disease and in experimental brain injuries (Liu et al., 2012; Hijioka et al., 2012). However, it is currently unclear whether this involves the modulation of microglia or astrocyte function, or whether α7nAChRs' actions are independent of its anti-inflammatory actions. Clearly, more investigations need to be done to understand direct and indirect effects of the cholinergic system on glia functions.
Q: What about high-pathology controls—30 percent of aged, non-demented individuals who have brains full of pathology?
Terrence Town: High-pathology controls are a terribly interesting cohort. However, there is an alternate interpretation for these individuals. Perhaps they have certain protective alleles that, despite the presence of AD-type neuropathological lesions, guard against cognitive decline. I think it would be extremely useful to apply genomewide approaches in this interesting group to determine exactly which modifier genes play a role in protection against dementia.
Irene Knuesel: I fully agree that these individuals are most fascinating because of their—obvious—neuroprotective/repair/defense strategies during brain aging.
References
News Citations
- Naproxen/Rofecoxib Trial Results Published
- NSAIDs in AD: Epi and Trial Data at Odds—Again
- Curbing Cell Cycle Re-entry: Window of Opportunity for NSAIDs?
- Vienna: In Genetics, Bigger Is Better—Data Sharing Nets Three New Hits
- Enter the New Alzheimer’s Gene: TREM2 Variant Triples Risk
- Macrophages Storm Blood-brain Barrier, Clear Plaques—or Do They?
- Soothing Neuroinflammation Quells Plaques in Mice
- Can Phagocytosis, Memory Effects Revive Diabetes Meds?
- DC: Is Alzheimer's Rooted in the Early Life?
- The Brain Minus Microglia—No Effect on Plaques
Paper Citations
- Breitner JC, Baker LD, Montine TJ, Meinert CL, Lyketsos CG, Ashe KH, Brandt J, Craft S, Evans DE, Green RC, Ismail MS, Martin BK, Mullan MJ, Sabbagh M, Tariot PN, . Extended results of the Alzheimer's disease anti-inflammatory prevention trial. Alzheimers Dement. 2011 Jul;7(4):402-11. PubMed.
- Meyer U, Nyffeler M, Engler A, Urwyler A, Schedlowski M, Knuesel I, Yee BK, Feldon J. The time of prenatal immune challenge determines the specificity of inflammation-mediated brain and behavioral pathology. J Neurosci. 2006 May 3;26(18):4752-62. PubMed.
- Meyer U, Engler A, Weber L, Schedlowski M, Feldon J. Preliminary evidence for a modulation of fetal dopaminergic development by maternal immune activation during pregnancy. Neuroscience. 2008 Jun 23;154(2):701-9. PubMed.
- Krstic D, Madhusudan A, Doehner J, Vogel P, Notter T, Imhof C, Manalastas A, Hilfiker M, Pfister S, Schwerdel C, Riether C, Meyer U, Knuesel I. Systemic immune challenges trigger and drive Alzheimer-like neuropathology in mice. J Neuroinflammation. 2012;9:151. PubMed.
- Streit WJ, Braak H, Xue QS, Bechmann I. Dystrophic (senescent) rather than activated microglial cells are associated with tau pathology and likely precede neurodegeneration in Alzheimer's disease. Acta Neuropathol. 2009 Oct;118(4):475-85. PubMed.
- Flanary BE, Sammons NW, Nguyen C, Walker D, Streit WJ. Evidence that aging and amyloid promote microglial cell senescence. Rejuvenation Res. 2007 Mar;10(1):61-74. PubMed.
- Stone JR, Singleton RH, Povlishock JT. Intra-axonal neurofilament compaction does not evoke local axonal swelling in all traumatically injured axons. Exp Neurol. 2001 Dec;172(2):320-31. PubMed.
- Smith DH, Chen XH, Iwata A, Graham DI. Amyloid beta accumulation in axons after traumatic brain injury in humans. J Neurosurg. 2003 May;98(5):1072-7. PubMed.
- Sundström A, Nilsson LG, Cruts M, Adolfsson R, Van Broeckhoven C, Nyberg L. Increased risk of dementia following mild head injury for carriers but not for non-carriers of the APOE epsilon4 allele. Int Psychogeriatr. 2007 Feb;19(1):159-65. PubMed.
- Grathwohl SA, Kälin RE, Bolmont T, Prokop S, Winkelmann G, Kaeser SA, Odenthal J, Radde R, Eldh T, Gandy S, Aguzzi A, Staufenbiel M, Mathews PM, Wolburg H, Heppner FL, Jucker M. Formation and maintenance of Alzheimer's disease beta-amyloid plaques in the absence of microglia. Nat Neurosci. 2009 Nov;12(11):1361-3. PubMed.
- Radde R, Bolmont T, Kaeser SA, Coomaraswamy J, Lindau D, Stoltze L, Calhoun ME, Jäggi F, Wolburg H, Gengler S, Haass C, Ghetti B, Czech C, Hölscher C, Mathews PM, Jucker M. Abeta42-driven cerebral amyloidosis in transgenic mice reveals early and robust pathology. EMBO Rep. 2006 Sep;7(9):940-6. PubMed.
- Choi SH, Veeraraghavalu K, Lazarov O, Marler S, Ransohoff RM, Ramirez JM, Sisodia SS. Non-cell-autonomous effects of presenilin 1 variants on enrichment-mediated hippocampal progenitor cell proliferation and differentiation. Neuron. 2008 Aug 28;59(4):568-80. PubMed.
- Vom Berg J, Prokop S, Miller KR, Obst J, Kälin RE, Lopategui-Cabezas I, Wegner A, Mair F, Schipke CG, Peters O, Winter Y, Becher B, Heppner FL. Inhibition of IL-12/IL-23 signaling reduces Alzheimer's disease-like pathology and cognitive decline. Nat Med. 2012 Dec;18(12):1812-9. PubMed.
- Wang H, Yu M, Ochani M, Amella CA, Tanovic M, Susarla S, Li JH, Yang H, Ulloa L, Al-Abed Y, Czura CJ, Tracey KJ. Nicotinic acetylcholine receptor alpha7 subunit is an essential regulator of inflammation. Nature. 2003 Jan 23;421(6921):384-8. PubMed.
- Liu Y, Hu J, Wu J, Zhu C, Hui Y, Han Y, Huang Z, Ellsworth K, Fan W. α7 nicotinic acetylcholine receptor-mediated neuroprotection against dopaminergic neuron loss in an MPTP mouse model via inhibition of astrocyte activation. J Neuroinflammation. 2012;9:98. PubMed.
- Hijioka M, Matsushita H, Ishibashi H, Hisatsune A, Isohama Y, Katsuki H. α7 Nicotinic acetylcholine receptor agonist attenuates neuropathological changes associated with intracerebral hemorrhage in mice. Neuroscience. 2012 Oct 11;222:10-9. PubMed.
Other Citations
External Citations
Further Reading
Papers
- True HL, Lindquist SL. A yeast prion provides a mechanism for genetic variation and phenotypic diversity. Nature. 2000 Sep 28;407(6803):477-83. PubMed.
Panelists
-
 Irene Knuesel
Roche
Irene Knuesel
Roche
-
 John Breitner, MD, MPH
McGill University Faculty of Medicine
John Breitner, MD, MPH
McGill University Faculty of Medicine
-
 Michael Heneka, M.D.
Klinik und Poliklinik für Neurologie
Michael Heneka, M.D.
Klinik und Poliklinik für Neurologie
-
 Frank Heppner
Charité Universitaetsmedizin Berlin
Frank Heppner
Charité Universitaetsmedizin Berlin
-
 Terrence Town, Ph.D.
University of Southern California
Terrence Town, Ph.D.
University of Southern California
Comments
Vrije Universiteit
Irene Knuesel's hypothesis that late-onset AD might be the result of a two-hit immune challenge is most interesting. Her strategy to develop a wild-type AD animal model is based on a sound scientific background. The genetic evidence for the role of innate immunity received strong support a few weeks ago when TREM2 variants were found to be risk factors for the disease. There is also epidemiological and clinical evidence that systemic inflammatory mediators can contribute to the clinical expression of late-onset AD (Eikelenboom et al., 2012). Overall, the research seems to suggest that innate immunity is a predisposing factor for late-onset AD, as is a second immune challenge in the elderly.
Neuropathological and experimental studies indicate a role for the innate immunity-related CD-14 and Toll-like receptor signaling pathways of glia cells in promoting proinflammatory cytokine production. These pathways are under genetic control, and offspring with a parental history of late-onset AD produce more proinflammatory cytokines (Van Exel et al., 2009). Patients with late-onset AD also have high comorbidities in contrast to those with early onset AD. These comorbidities are frequently accompanied with systemic or local inflammation. It will be interesting to know whether and how peripheral inflammation interacts with glial pathways to increase proinflammatory cytokine production within the brain. The animal model developed by Knuesel and colleagues shows promise for unraveling the precise mechanisms.
Next to the experimental animal work, clinical studies should be performed to answer the question, What makes older patients with peripheral inflammation so vulnerable for late-onset AD? Most interesting in this regard is current research on the role of inflammation in delirium. For example, there is increasing evidence that postoperative delirium after hip surgery is an important predictor of incident dementia in elderly patients living independently at home. Contrary to popular belief, there is little evidence that general anesthesia is associated with delirium after surgery. In time course studies in older patients admitted for surgery after hip fracture, significant differences in serum levels of interleukin-6 and C-reactive protein were found between patients with and without delirium (van Munster et al., 2008). The release of these inflammatory mediators seems to be a consequence of the fracture and the tissue destruction resulting from surgery. In particular, clinical trials are warranted in selected patient samples, such as elderly patients undergoing hip surgery, to investigate whether anti-inflammatory drugs can prevent further cognitive decline in patients with mild cognitive impairment whenever peripheral inflammation exerts increased inflammatory pressure on the brain.
References:
Eikelenboom P, Hoozemans JJ, Veerhuis R, van Exel E, Rozemuller AJ, Van Gool WA. Whether, when and how chronic inflammation increases the risk of developing late-onset Alzheimer's disease. Alzheimers Res Ther. 2012;4(3):15. PubMed.
van Exel E, Eikelenboom P, Comijs H, Frölich M, Smit JH, Stek ML, Scheltens P, Eefsting JE, Westendorp RG. Vascular factors and markers of inflammation in offspring with a parental history of late-onset Alzheimer disease. Arch Gen Psychiatry. 2009 Nov;66(11):1263-70. PubMed.
van Munster BC, Korevaar JC, Zwinderman AH, Levi M, Wiersinga WJ, de Rooij SE. Time-course of cytokines during delirium in elderly patients with hip fractures. J Am Geriatr Soc. 2008 Sep;56(9):1704-9. PubMed.
�
The potential benefit of using an NSAID or NSAID-like compound will be best seen when testing those high-risk/genetically enriched young asymptomatic subjects, such as ApoE4 homozygotes/heterozygotes in a clinical trial.
Before cognitive symptoms are noticed by the individual or a well-experienced psychometrician/neuropsychologist, the neuroinflammatory cascade is well underway. It is likely that activation of microglia class M1 and inhibition of the phagocytic class of microglia, M2, are in play at the asymptomatic stage.
As has been eloquently presented by the panelists on the Webinar, this may be the only or the "best/ideal" window of opportunity to slow progression in asymptomatic individuals to symptomatic mild cognitive impairment/dementia.
I and my colleagues hope to test a compound that might stimulate phagocytic M2 microglia at this early phase, and possibly inhibit the activated M1 microglia subtype (see ARF related news story). Only by starting this type of study very early in the pathogenesis of AD can modulation of the neuroimmune responses to the many well-described risk factors for AD be shown to be effective.
References:
Richter-Schmidinger T, Alexopoulos P, Horn M, Maus S, Reichel M, Rhein C, Lewczuk P, Sidiropoulos C, Kneib T, Perneczky R, Doerfler A, Kornhuber J. Influence of brain-derived neurotrophic-factor and apolipoprotein E genetic variants on hippocampal volume and memory performance in healthy young adults. J Neural Transm. 2011 Feb;118(2):249-57. PubMed.
Maezawa I, Jenkins DP, Jin BE, Wulff H. Microglial KCa3.1 Channels as a Potential Therapeutic Target for Alzheimer's Disease. Int J Alzheimers Dis. 2012;2012:868972. PubMed.
If you are interested in seeing/hearing more on the inflammation-axonopathy model of AD by Irene and me, see this European Journal of Neuroscience interview.
Any feedback, comments, and/or criticism are highly appreciated.
Make a Comment
To make a comment you must login or register.