Protective APOE3 Variant Binds More Lipids, Self-Aggregates Less
Quick Links
A rare APOE3 variant, dubbed APOE3-Jacksonville, drastically reduces a person's risk of developing Alzheimer’s disease. How does it protect the brain? Primarily by reducing ApoE self-aggregation, according to scientists led by Guojun Bu at the Mayo Clinic. In the September 29 Science Translational Medicine, co-first authors Chia-Chen Liu, Melissa Murray, Xia Li, and Na Zhao reported that the APOE3-Jac variant creates a protein that can pack on the fat. This increased the amount of healthy lipids in the brains of mice and reduced ApoE self-aggregation. In a mouse model of amyloidosis, APOE3-Jac triggered a cascade of fewer plaques and fewer dystrophic neurites, hinting that ApoE's lipidation or self-aggregation may become a therapeutic target.
- APOE3-Jacksonville variant causes missense mutation in the lipid-binding region.
- 5xFAD mice expressing the variant had fewer plaques, less neuron damage.
- Could reducing ApoE aggregation be a therapeutic opportunity?
“This paper emphasizes the importance of lipid metabolism in the context of AD,” Mikko Hiltunen, University of Eastern Finland, Kuopio, told Alzforum. William Rebeck, Georgetown University, Washington, D.C., agreed. "It clarifies the models of APOE-related risk and convincingly suggests ApoE lipidation as a good therapeutic target to reduce neurodegeneration risk," he wrote (comment below).

All Aboard the Shuttle. Model of a cholesterol efflux assay. Astrocytes are cultured with ApoE3 or ApoE3-Jac (blue crescent). Scientists assess ApoE’s lipid-binding capacity by measuring the amount of cholesterol (yellow circles) released in the form of lipoprotein particles (top left). [Courtesy of Liu et al., Science Translational Medicine, 2021.]
Previously, Bu and colleagues found the APOE3-Jac variant in people who had remained mentally sharp into very old age (Medway et al., 2014). To find out why, the scientists collected frontal cortex tissue in the Mayo Clinic Brain Bank from 1,128 controls, 457 people who had had AD, and 1,063 people who had had dementia with Lewy bodies. Sequencing confirmed that five controls—and no cases—carried the APOE3-Jac variant.
The APOE3-Jac mutation, V236E, lies in ApoE's C-terminal and lipid-binding region. Could carriers benefit by way of altered brain lipid metabolism? To find out, the scientists immunoprecipitated ApoE from postmortem brain tissue of three E3/E3-Jac carriers and five E3/E3 controls. ApoE from carriers bound more cholesterol, hinting that it could better haul lipids. The researchers quantified the mutant protein’s lipid-loading capacity by isolating astrocytes, the main ApoE-producing cells, from APOE knockout mice and culturing them with purified human ApoE3 or ApoE3-Jac (see image above). Indeed, the Jac mutant shuttled more cholesterol out of the cells (image below).
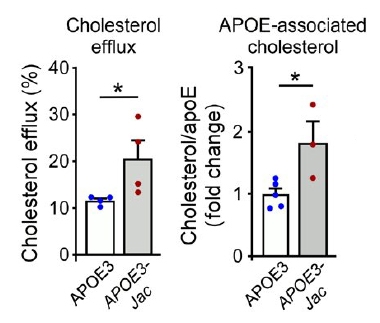
Fat and Functional. In cultured astrocytes (left), ApoE3-Jac pumped more cholesterol into the medium (gray bar). In human frontal cortex tissue preps (right), ApoE3-Jac bound more cholesterol (gray bar). [Courtesy of Liu et al., Science Translational Medicine, 2021.]
This finding made Liu and colleagues curious about lipids other than cholesterol. They injected a virus encoding APOE3 or APOE3-Jac into brain ventricles of APOE knockout mice, and later found that brain tissue from Jac-expressing mice contained not only more cholesterol but also more lipids crucial to membrane homeostasis and synaptic function, including phosphatidylserine, phosphatidylethanolamine, phosphatidic acid, sulfatide, and cerebroside. "Alterations in lipid metabolism likely contribute to the protective effects of APOE3-Jac," Jacob Raber, Oregon Health & Science University, Portland, wrote to Alzforum (comment below).
If lipids grease up ApoE3-Jac, might that make the protein less likely to stick to itself? The scientists fractionated human brain tissue, and found that while ApoE3-Jac carriers and controls had comparable amounts of soluble ApoE, carriers had much less insoluble ApoE, suggesting fewer aggregates. Indeed, in vitro, ApoE3 readily oligomerized and bound heparin, whereas ApoE3-Jac was primarily monomeric and bound heparin poorly. "Impaired interaction with heparan-sulphate proteoglycans may be a common feature of APOE variants associated with reduced dementia risk, including APOE3-Christchurch and APOE2," Yakeel Quiroz, Massachusetts General Hospital, Boston, wrote (comment below).
ApoE is known to fuel Aβ aggregation, hence the scientists wondered if the Jac variant’s aversion to self-aggregation would carry over to amyloid deposition. In brain tissue from Jac carriers, they found less soluble Aβ42, less insoluble Aβ42 and Aβ40, and few or no diffuse plaques. On PET scans in living people, they saw less amyloid and greater glucose utilization in a carrier than controls (image below).
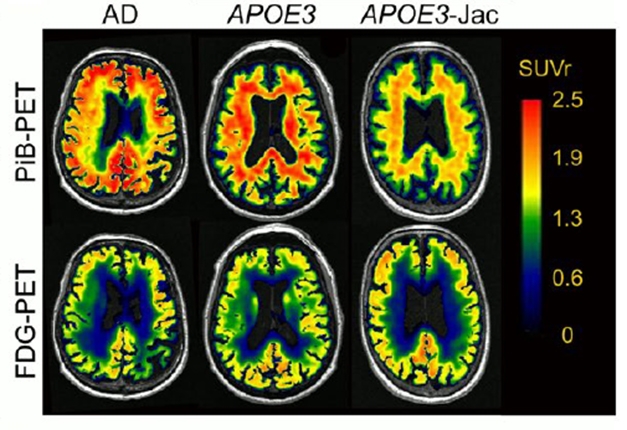
A Healthier Brain. An aged APOE3-Jac carrier's brain (right) had less amyloid (top) and metabolized more glucose (bottom) than that of an aged cognitively normal APOE3 homozygote (middle) or a person with AD (left). [Courtesy of Liu et al., Science Translational Medicine, 2021.]
APOE genotype is a major risk factor for both AD and DLB, hinting that ApoE self-aggregation may be involved in aberrant protein aggregation in both types of dementia. “While this paper focuses on the levels and deposition of Aβ, the observations that APOE4 increases risk of DLB while APOE3-Jac is protective suggest that important APOE-related processes are not limited to APOE-Aβ interactions,” Rebeck wrote.
To explore APOE3-Jac's effect on amyloid deposition and its consequences, Liu and colleagues virally expressed APOE3 and APOE3-Jac in 5xFAD amyloidosis mice. Besides recapitulating the finding of fewer cortical plaques in APOE3-Jac-expressing mice, this experiment also showed them to have fewer microglia, reactive astrocytes, and dystrophic neurites (image below). "This emphasizes the critical importance of ApoE structure on AD-related processes," David Holtzman, Washington University, St. Louis, wrote to Alzforum (comment below).
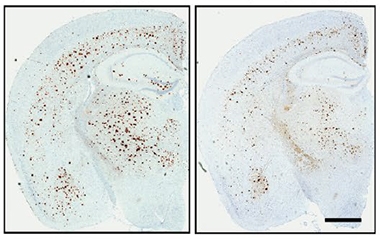
Jac Means Less Plaque. Four-month-old 5xFAD mice expressing APOE3-Jac (right) had fewer cortical plaques than mice expressing APOE3 (left). [Courtesy of Liu et al., Science Translational Medicine, 2021.]
However, APOE3-Jac mice performed similarly to APOE3-expressing 5xFAD mice on memory tasks. Commentators were curious if APOE3-Jac also affects tau pathology, and whether it may affect the memories of mice with plaques and tangles. “These studies definitely need to be extended to an animal model that includes tau pathology, as the 5xFAD used here is a limitation,” Ralph Martins, Edith Cowan University, Australia, wrote to Alzforum (full comment below). Together with the Jackson Laboratory in Bar Harbor, Maine, Bu and colleagues are creating APOE3-Jac knock-in mouse models on different backgrounds to define the variant’s effect on cognition and AD pathology.
Regarding therapy development, Jing Guo, Nga Bien-Ly, and Gilbert Di Paolo at Denali Therapeutics, San Francisco, noted that previous strategies to reduce ApoE aggregation focused on increasing its lipidation via the LXR/RXR lipid receptor pathways or boosting the cholesterol transporter ABCA1 (full comment below). Bu and colleagues focus on reducing ApoE aggregation itself. “Monomeric ApoE is a much better lipid acceptor than aggregated ApoE,” he told Alzforum. His group is screening FDA-approved, brain-penetrating molecules and is working on structure-based drug design in search of new compounds that disrupt ApoE aggregation.
APOE3-Jac is rare, and its frequency needs to be better defined in larger cohorts. Within a Danish cohort of more than 100,000 people, geneticists led by Ruth Frikke‐Schmidt, University of Copenhagen, found a lower frequency of this variant than what Liu and colleagues had reported. APOE3-Jac carriers did have a 70 percent lower dementia risk, albeit just short of statistical significance, (Rasmussen et al., 2020). Alison Goate and Anna Podlesny-Drabiniok, Icahn School of Medicine at Mount Sinai, New York, noted an even lower frequency of APOE3-Jac among almost 100,000 people in the gnomAD database. Nancy Ip, Hong Kong University of Science and Technology, who studies the ApoE genetic locus, also called for determining APOE3-Jac 's frequency across ethnic groups (comments below).—Chelsea Weidman Burke
References
Research Models Citations
Paper Citations
- Medway CW, Abdul-Hay S, Mims T, Ma L, Bisceglio G, Zou F, Pankratz S, Sando SB, Aasly JO, Barcikowska M, Siuda J, Wszolek ZK, Ross OA, Carrasquillo M, Dickson DW, Graff-Radford N, Petersen RC, Ertekin-Taner N, Morgan K, Bu G, Younkin SG. ApoE variant p.V236E is associated with markedly reduced risk of Alzheimer's disease. Mol Neurodegener. 2014 Mar 10;9:11. PubMed.
- Rasmussen KL, Tybjaerg-Hansen A, Nordestgaard BG, Frikke-Schmidt R. APOE and dementia - resequencing and genotyping in 105,597 individuals. Alzheimers Dement. 2020 Dec;16(12):1624-1637. Epub 2020 Aug 18 PubMed.
External Citations
Further Reading
Papers
- Rasmussen KL, Tybjaerg-Hansen A, Nordestgaard BG, Frikke-Schmidt R. APOE and dementia - resequencing and genotyping in 105,597 individuals. Alzheimers Dement. 2020 Dec;16(12):1624-1637. Epub 2020 Aug 18 PubMed.
Primary Papers
- Liu CC, Murray ME, Li X, Zhao N, Wang N, Heckman MG, Shue F, Martens Y, Li Y, Raulin AC, Rosenberg CL, Doss SV, Zhao J, Wren MC, Jia L, Ren Y, Ikezu TC, Lu W, Fu Y, Caulfield T, Trottier ZA, Knight J, Chen Y, Linares C, Wang X, Kurti A, Asmann YW, Wszolek ZK, Smith GE, Vemuri P, Kantarci K, Knopman DS, Lowe VJ, Jack CR Jr, Parisi JE, Ferman TJ, Boeve BF, Graff-Radford NR, Petersen RC, Younkin SG, Fryer JD, Wang H, Han X, Frieden C, Dickson DW, Ross OA, Bu G. APOE3-Jacksonville (V236E) variant reduces self-aggregation and risk of dementia. Sci Transl Med. 2021 Sep 29;13(613):eabc9375. PubMed.
Annotate
To make an annotation you must Login or Register.
Comments
Georgetown University
This study is a dramatic demonstration of APOE characteristics that promote neurodegeneration, greatly clarifying the more general models of APOE-related risk. In convincing ways, it pushes forward ideas from disparate studies to demonstrate that APOE lipidation is a good therapeutic target for reducing the risk of neurodegeneration.
The authors find that APOE3-Jac increases APOE lipidation; this is consistent with data showing that APOE4 has lower levels of lipidation than APOE3 in cell culture models, in mouse models, and in human CSF.
APOE3-Jac increases lipid efflux from cells compared to APOE3, consistent with cell culture findings that cholesterol efflux models the protective nature of the variants APOE2>APOE3>APOE4.
APOE3-Jac has less self-aggregates than APOE3 that occur through the lipid-binding domain, while APOE4 has more. APOE3-Jac reduces the amount of microglial responses to Aβ deposits, consistent with findings from human APOE mouse models comparing APOE3 and APOE4.
Although some of this new work on APOE3-Jac focuses on the effects on levels and deposition of Aβ, the observations that APOE4 increases risk of diffuse Lewy body disease and that APOE3-Jac is protective in DLB suggest that the important APOE-related processes are not limited to APOE-Aβ interactions.
OHSU
This is a tour de force study. Liu et al. compared the effects of APOE3 with the APOE3-Jacksonville variant associated with healthy brain aging and reduced risk for AD and dementia with Lewy bodies (DLB), on aggregation of ApoE, lipidation, and alterations in lipids in brain important for synaptic function. These data nicely supplement the results seen in an individual with the APOE3 Christchurch mutation, which protected against autosomal-dominant AD, and individuals with the protective variant of the human amyloid precursor protein (APP) revealed in an Icelandic population (Jonsson et al., 2012).
Antemortem and postmortem analysis showed a lack of global atrophy, amyloid pathology, in the oldest-old carrying the V236E variant. The V236E variant was not revealed in patients with dementia with Lewy Bodies either. The cholesterol-to-ApoE ratio was higher in brains of individuals with the V236E variant. The amount of soluble ApoE was similar in individuals with APOE3 and the V236E variant. However, those carrying the V236E variant had less insoluble ApoE, less soluble Aβ42, and less insoluble Aβ40 and Aβ42, only a few diffuse amyloid plaques or no plaque pathology at all.
In astrocytes derived from Apoe-/- mice, the V236E variant was more efficient in promoting cholesterol efflux. Alterations in lipid metabolism are likely important in these protective effects.
When AAV8-expressing APOE3 or the V236E variant under the control of astrocyte-specific GFAP promoter were bilaterally injected into the cerebral lateral ventricles of mice at postnatal day 2, modules that comprise cholesterol, cholesterol ester, phosphatidic acid (PA), phosphatidylserine (PS), phosphatidylethanolamine (PE), sulfatide, and cerebroside were positively correlated with expression of the V236E variant. These data support brain lipids as important therapeutic targets in AD.
Using the same approach in a 5xFAD amyloid mouse model, reduced amyloid pathology in the cortex and reduced levels of insoluble Aβ40 and Aβ42, less reactive astrocytes, less dystrophic neurites around plaques, were seen. The V236E variant did not affect accumulation of mouse ApoE in the amyloid plaques, the amount of disease-associated microglia around plaques or immunoreactivity of CD68, a marker of activated microglia, or activity levels or measures of anxiety in the open field or elevated plus maze or freezing in the hippocampus-dependent contextual or hippocampus-independent cued fear memory tests in 4-month-old 5-FAD mice.
Based on the reduced risk for AD and DLB in those with the V236E variant, the lack of behavioral or cognitive effects in light of all neuropathological changes seen in the brains of the mice in this study is important to note. As we reported, there are isoform-dependent effects of ApoE on spatial-memory retention in the water maze in 6-month-old mice lacking mouse ApoE and expressing human APP with two AD mutations, prior to the onset of plaque pathology and in the absence of genotype differences in hippocampal or cortical Aβ levels (Raber et al., 2000). One would expect to see behavioral and cognitive changes in the presence of the neuropathological changes seen in the current study. It is important to note that in the mouse model used in the current study, both human and mouse APP and ApoE present, which might complicate the interpretation of the results. Behavioral alterations and impaired cognitive performance might have been revealed in mice only containing human APP and ApoE and lacking their murine counterparts. It is also possible that behavioral alterations and impaired cognitive performance might be revealed in older mice, but it still questions the exact role of the neuropathological changes seen in behavioral and cognitive performance. This is very important to address in future studies considering the recent approval for using antibodies clearing Aβ for AD patients in the absence of convincing beneficial cognitive effects yet (Selkoe, 2021).
It is conceivable that the protective effects of the V236E variant on behavioral and cognitive performance might involve additional pathways to the ones revealed in this study. For example, recently it was shown that, in centenarians, there were protective variants in genes involved in insulin and AMP-activating protein kinase (AMPK) signaling. In centenarian E4 carriers, there were functional variants in the Wnt signaling pathway associated with extended life, suggesting that this pathway might protect against detrimental effects of carrying E4 (Lin et al., 2021).
Increased understanding of pathways protecting against AD in individuals with distinct APOE genotypes will likely facilitate the development of novel therapeutic strategies to reduce age-related cognitive decline and cognitive injury in AD and related neurodegenerative diseases.
References:
Jonsson T, Atwal JK, Steinberg S, Snaedal J, Jonsson PV, Bjornsson S, Stefansson H, Sulem P, Gudbjartsson D, Maloney J, Hoyte K, Gustafson A, Liu Y, Lu Y, Bhangale T, Graham RR, Huttenlocher J, Bjornsdottir G, Andreassen OA, Jönsson EG, Palotie A, Behrens TW, Magnusson OT, Kong A, Thorsteinsdottir U, Watts RJ, Stefansson K. A mutation in APP protects against Alzheimer's disease and age-related cognitive decline. Nature. 2012 Aug 2;488(7409):96-9. PubMed.
Raber J, Wong D, Yu GQ, Buttini M, Mahley RW, Pitas RE, Mucke L. Apolipoprotein E and cognitive performance. Nature. 2000 Mar 23;404(6776):352-4. PubMed.
Selkoe DJ. Treatments for Alzheimer's disease emerge. Science. 2021 Aug 6;373(6555):624-626. PubMed.
Lin JR, Sin-Chan P, Napolioni V, Zhang ZD. Rare genetic coding variants associated with human longevity and protection against age-related diseases. Nature Aging, 1, 2021, pp.783–94. Nat Aging.
Massachusetts General Hospital, Harvard Medical School
Patients with resistance to Alzheimer’s disease and resilience to cognitive decline are leading a revolution in our understanding of Alzheimer’s pathophysiology and paving the way for new, patient-inspired therapies. This interesting piece from Dr. Bu’s laboratory explores the mechanisms via which the APOE3-Jacksonville (V236E) variant reduces dementia risk. Their focus on the effects on amyloid is justified by findings suggesting significantly reduced amyloid pathology in the presence of the APOE3-Jacksonville. It remains to be established to which extent APOE3-Jacksonville impacts tau pathology, which was greatly reduced in an individual my group previously reported with three-decades-long resistance to autosomal-dominant Alzheimer’s disease who was homozygous for the APOE3-Christchurch variant.
In terms of molecular mechanism, Dr. Bu’s group focused on the effects of the V236E variant on APOE self-aggregation, a feature previously associated with APOE4 toxicity. Interestingly, they also reported that APOE3-Jacksonville, like APOE3-Christchurch, reduces interactions of ApoE with heparin, a glycosaminoglycan. It appears that impaired interaction with heparan-sulphate proteoglycans (HSPGs) may be a feature common to APOE variants associated with reduced risk to dementia, including APOE3-Christchurch, APOE3-Jacksonville, and APOE2.
This impaired interaction with HSPGs may result from direct changes to the HSPG binding domain like in Christchurch, or indirectly, as proposed by the authors, as a result of reduced APOE oligomerization as in APOE3-Jacksonville.
Impaired interaction with lipoprotein receptors does not appear to be a common mechanism, as it is reduced in Christchurch and APOE2 but preserved for APOE3-Jacksonville. A focus on common mechanisms can definitely aid the field to develop new and effective therapies for Alzheimer’s disease.
Washington University
This interesting study assessed the effects of the ApoE3 V236E Jacksonville variant in several ways. Previous studies have suggested that the presence of this rare variant decreases risk of AD to a similar extent as APOE2. Here, the authors further confirm that association but they also assess the potential mechanisms leading to the effect.
Interestingly, when the properties of APOE3-V236E vs. APOE3 were compared, the APOE3-V236E was clearly less prone to aggregate. In addition, the variant was associated with increased cholesterol efflux. The effects of the APOE3-V236E variant were assessed in a mouse model of amyloidosis and showed that it resulted in less amyloid deposition, less neuritic dystrophy, less microglia, and less ApoE in plaques.
While the mechanism(s) as to how ApoE contributes to ApoE and AD are likely pleiotropic, one way that it definitely contributes to AD is via influencing Aβ aggregation and its downstream effects. This interesting study suggests the possibility that slight alterations in ApoE structure resulting in properties such as altered aggregation propensity, may be influencing Aβ aggregation as well as potentially other effects in the brain such as cholesterol efflux and specific effects on lipid metabolism. This study emphasizes the critical importance of ApoE structure in the effects of ApoE on critical disease-related processes.
Denali Therapeutics
Denali Therapeutics
Denali Therapeutics
For more than 25 years, the AD field has established that APOE is the strongest genetic risk factor for late-onset AD (LOAD), with the APOE2 allele conferring the lowest risk and the APOE4 allele the greatest risk compared to the APOE3 allele (e.g., Long and Holtzman, 2019). In addition to these relatively common variants, there are various rare mutants of APOE that appear to modify AD risk. They are increasingly being investigated in the hope of identifying the molecular mechanisms modulating this risk as well as potential novel therapeutics.
For instance, it was first reported in 2014 that the APOE3-Jac mutant V236E was associated with a reduction in LOAD risk that is perhaps even more dramatic than that observed for APOE2 (Medway et al., 2014). The exact protective mechanism has remained enigmatic until this new paper by Liu et al., which has now shed some light on how the V236E mutation could decrease the risk for AD by reducing aggregation of ApoE protein.
It was previously established that the C-terminal region of ApoE, where the V236E mutation resides, is prone to self-aggregation and is also critically involved in lipid binding. In this study, ApoE3-Jac and ApoE4-Jac secreted by HEK293 cells, a heterologous system, were found to exist predominantly in the form of monomers, whereas ApoE3 and ApoE4 were secreted primarily as oligomers, suggesting that the V236E mutation decreases the aggregation propensity of ApoE.
Moreover, the authors demonstrated that ApoE3-Jac and ApoE4-Jac led to more efficient cholesterol efflux than ApoE3 and ApoE4, respectively. In 5xFAD mice with AAV-mediated ApoE overexpression under the GFAP promoter, ApoE3-Jac expression resulted in lower fibrillar plaque load and reduced ApoE associated with plaques, accompanied by reduced astrogliosis and dystrophic neurites, compared to ApoE3 expression. ApoE3-Jac overexpression in the mouse brain also caused subtle changes in some lipid species, including various phospholipids and myelin lipids (e.g., sulfatides), although the significance of these lipid changes is unclear.
Results from the in vivo study recapitulated at least in part findings from the limited human brains of APOE3-Jac carriers available, where the authors reported greatly reduced insoluble ApoE and a lower burden of plaques with more diffuse and amorphous morphology.
This elegantly designed and thoroughly conducted study has built a compelling case for the aggregation-reducing effect of the V236E mutation being a key mechanism mediating its AD protection. It has long been suggested that increased aggregation propensity of ApoE4 is at least partly responsible for its deleterious role in promoting AD risk (Yamazaki et al., 2019). The correlation between self-aggregation of ApoE and amyloid plaque burden, together with frequent deposition of ApoE at the core of plaques, strongly suggests ApoE could be directly seeding plaque formation in AD brains. This is in line with our previous study, in collaboration with the Holtzman lab, demonstrating plaque reduction in the APPPS1-21/APOE4 mice by a monoclonal antibody specifically targeting poorly lipidated, aggregated ApoE (Liao et al., 2018).
As already hypothesized in the field, a “structural corrector” capable of reducing ApoE4 aggregation may represent a promising therapeutic approach for APOE4 carriers, although it may be quite challenging to develop small molecules that can inhibit protein aggregation with high specificity. An alternative strategy to reduce ApoE aggregation is to increase its lipidation state, as suggested by studies in the field on nuclear receptors (e.g., LXR, RXR) as well as ABCA1, the main cholesterol transporter in the CNS (Lanfranco et al., 2020).
As discussed by Liu et al, there appears to be an inverse relationship between ApoE aggregation and lipidation, whereby augmented lipidation of ApoE may abate its aggregation, and vice versa. Intriguingly, V236E was shown to promote cholesterol efflux and thus ApoE lipidation, which could further decrease its propensity for self-aggregation, giving rise to the most favorable ApoE species (i.e., highly lipidated, not aggregated) circulating in the brain. The protective effect of increased lipidation of ApoE is also consistent with the identification of additional LOAD risk genes involved in lipid efflux, including ABCA1 and ABCA7, the genetic deletion of which has been shown to increase plaque deposition in mouse brains (Dib et al., 2021; Lanfranco et al., 2020).
Whether the APOE3-Jac variant is exclusively protective against amyloid deposition, or whether it can also decrease tau pathology, remains to be determined. For example, given the link between cholesterol accumulation and tau pathology observed in Niemann-Pick disease type C, enhanced cholesterol efflux may have a beneficial effect of reducing tau aggregation by lowering intracellular cholesterol load, although the exact mechanism is not clearly understood.
Moreover, the authors described reduced heparin binding of the ApoE3-Jac variant, which was attributed mostly to a loss of avidity in binding due to its monomeric form. Importantly, a 2019 study reported impaired heparin binding of the ApoE3-Christchurch mutant, which was found to be associated with highly restricted tau pathology despite abundant amyloid plaques in one PSEN1 mutation carrier (Arboleda-Velasquez et al., 2019). Whether decreased ApoE binding to heparan sulfate proteoglycans (HSPGs) could be a protective mechanism shared by ApoE3-Jac, ApoE3-Christchurch and ApoE2 warrants further investigation. Specifically, cell surface HSPGs have been demonstrated to play a key role in mediating uptake of pathological tau seeds. ApoE-HSPG interactions may conceivably promote this process, leading to increased tau spreading in AD.
We look forward to seeing the plaque-reducing effect of the ApoE3-Jac variant replicated in an APP/ApoE3-Jac knock-in mouse model without the confound of endogenous mouse ApoE expression and with proper cell-autonomous expression of ApoE3-Jac in reactive microglia. In addition, functional characterization of ApoE3-Jac and ApoE4-Jac in iPSC-derived CNS cells, co-cultures and organoids will also be very informative.
References:
Arboleda-Velasquez JF, Lopera F, O'Hare M, Delgado-Tirado S, Marino C, Chmielewska N, Saez-Torres KL, Amarnani D, Schultz AP, Sperling RA, Leyton-Cifuentes D, Chen K, Baena A, Aguillon D, Rios-Romenets S, Giraldo M, Guzmán-Vélez E, Norton DJ, Pardilla-Delgado E, Artola A, Sanchez JS, Acosta-Uribe J, Lalli M, Kosik KS, Huentelman MJ, Zetterberg H, Blennow K, Reiman RA, Luo J, Chen Y, Thiyyagura P, Su Y, Jun GR, Naymik M, Gai X, Bootwalla M, Ji J, Shen L, Miller JB, Kim LA, Tariot PN, Johnson KA, Reiman EM, Quiroz YT. Resistance to autosomal dominant Alzheimer's disease in an APOE3 Christchurch homozygote: a case report. Nat Med. 2019 Nov;25(11):1680-1683. Epub 2019 Nov 4 PubMed.
Dib S, Pahnke J, Gosselet F. Role of ABCA7 in Human Health and in Alzheimer's Disease. Int J Mol Sci. 2021 Apr 27;22(9) PubMed.
Lanfranco MF, Ng CA, Rebeck GW. ApoE Lipidation as a Therapeutic Target in Alzheimer's Disease. Int J Mol Sci. 2020 Sep 1;21(17) PubMed.
Liao F, Li A, Xiong M, Bien-Ly N, Jiang H, Zhang Y, Finn MB, Hoyle R, Keyser J, Lefton KB, Robinson GO, Serrano JR, Silverman AP, Guo JL, Getz J, Henne K, Leyns CE, Gallardo G, Ulrich JD, Sullivan PM, Lerner EP, Hudry E, Sweeney ZK, Dennis MS, Hyman BT, Watts RJ, Holtzman DM. Targeting of nonlipidated, aggregated apoE with antibodies inhibits amyloid accumulation. J Clin Invest. 2018 May 1;128(5):2144-2155. Epub 2018 Mar 30 PubMed.
Long JM, Holtzman DM. Alzheimer Disease: An Update on Pathobiology and Treatment Strategies. Cell. 2019 Oct 3;179(2):312-339. Epub 2019 Sep 26 PubMed.
Medway CW, Abdul-Hay S, Mims T, Ma L, Bisceglio G, Zou F, Pankratz S, Sando SB, Aasly JO, Barcikowska M, Siuda J, Wszolek ZK, Ross OA, Carrasquillo M, Dickson DW, Graff-Radford N, Petersen RC, Ertekin-Taner N, Morgan K, Bu G, Younkin SG. ApoE variant p.V236E is associated with markedly reduced risk of Alzheimer's disease. Mol Neurodegener. 2014 Mar 10;9:11. PubMed.
Yamazaki Y, Zhao N, Caulfield TR, Liu CC, Bu G. Apolipoprotein E and Alzheimer disease: pathobiology and targeting strategies. Nat Rev Neurol. 2019 Sep;15(9):501-518. Epub 2019 Jul 31 PubMed.
University of Copenhagen
Increasing evidence from large-scale studies of the general population (Rasmussen et al., 2020) and from large pedigree family studies (Arboleda-Velasquez et al., 2019), suggests that structural variation in APOE beyond the well-known and common ɛ2/ɛ3/ɛ4 gene variants may play a role in dementia susceptibility.
Here, Liu et al. show that a structural APOE variant, Val236Glu (new nomenclature p.Val254Glu), with a population frequency of two to three per 1,000 individuals, functionally is associated with less self-aggregation and increased lipidation of the ApoE protein. These properties are suggested to explain the protective effect of the 236Glu form, as the variant in vitro ameliorates amyloid plaque-associated ApoE, plaque load, and related toxicity.
The author group previously observed that the 236Glu form was protective against AD dementia in a population from the Mayo Clinic in Jacksonville, and now substantiates these findings with mechanistic evidence. These findings are supported by a comprehensive sequencing and genotyping effort we conducted in prospective cohorts of 100,000 individuals from the Danish general population.
Collectively, when addressing susceptibility for dementia, it is now clear that the entire APOE locus should be addressed, as important structural variation beyond the ɛ2/ɛ3/ɛ4 APOE gene variants exists. The findings from Liu et al., as well as from Arboleda-Velasquez et al., and from our population sequencing efforts cited above, provide insights that may facilitate development of therapeutic strategies targeting ApoE aggregation and lipidation, receptor binding affinity, and ApoE concentrations.
References:
Rasmussen KL, Tybjaerg-Hansen A, Nordestgaard BG, Frikke-Schmidt R. APOE and dementia - resequencing and genotyping in 105,597 individuals. Alzheimers Dement. 2020 Dec;16(12):1624-1637. Epub 2020 Aug 18 PubMed.
Arboleda-Velasquez JF, Lopera F, O'Hare M, Delgado-Tirado S, Marino C, Chmielewska N, Saez-Torres KL, Amarnani D, Schultz AP, Sperling RA, Leyton-Cifuentes D, Chen K, Baena A, Aguillon D, Rios-Romenets S, Giraldo M, Guzmán-Vélez E, Norton DJ, Pardilla-Delgado E, Artola A, Sanchez JS, Acosta-Uribe J, Lalli M, Kosik KS, Huentelman MJ, Zetterberg H, Blennow K, Reiman RA, Luo J, Chen Y, Thiyyagura P, Su Y, Jun GR, Naymik M, Gai X, Bootwalla M, Ji J, Shen L, Miller JB, Kim LA, Tariot PN, Johnson KA, Reiman EM, Quiroz YT. Resistance to autosomal dominant Alzheimer's disease in an APOE3 Christchurch homozygote: a case report. Nat Med. 2019 Nov;25(11):1680-1683. Epub 2019 Nov 4 PubMed.
Icahn School of Medicine at Mount Sinai
This interesting and important paper uses human genetic data around a rare variant in APOE to direct functional studies in vivo and in vitro. These studies shed light on the role of APOE in Aβ deposition in AD and suggest that pharmacological targeting of the APOE C-terminal domain may be a novel therapeutic approach to AD treatment.
Using whole-exome sequencing (WES), the authors found a variant within APOE, in which valine is replaced by glutamate at position 236 (V236E); it appears to protect against AD and is associated with healthy brain aging. The cohort of AD cases (n=392) and cognitively normal controls (>85 years) (n=108) in this study was small, and the frequency of this allelic variant (APOE3-Jac) was 2 percent in controls.
However, replication studies using a larger cohort of controls (n=975) and diseased individuals (dementia with Lewy bodies, DLB n=1,063) showed even lower frequency of this allele (~0.3 percent) in controls, raising a question about the frequency of APOE3-Jac in larger cohorts with WGS/WES such as ADSP (Alzheimer’s disease sequencing project) or the Wellderly study.
Indeed, the frequency of this variant in the gnomAD database is 0.0009 in non-Finnish EUR and 0.00044 in AFR/African Americans based on close to 100,000 individuals. In addition, this variant seems not to be enriched in the elderly population (>80 years) based on data from 2,000 individuals (gnomAD), suggesting that replication of this genetic observation in larger cohorts will be needed to estimate the protective effect associated with the variant more precisely.
An interesting aspect of this paper is biochemical characteristics of APOE3-Jac both in vitro and in vivo demonstrating strong anti-aggregating propensities of APOE3-Jac and increased efficiency to promote reverse cholesterol transport in primary astrocytes. This suggests APOE3-Jac may facilitate lipid clearance processes in astrocytes and prevent formation of foamy glia frequently observed in APOE4 brains and iPSC-derived astrocytes and microglia with APOE4/4 genotype. (TCW et al., 2019; Lee et al., 2021).
In addition, generation of the animal model of amyloidosis (5xfAD) with APOE3-Jac expressed under the astrocytic GFAP promoter might be a useful tool to address astrocyte-microglial cross-talk in progression of AD pathology. Particularly, APOE3-Jac secreted from astrocytes may have an impact on microglia, which express the APOE3 isoform in this model, as they fail to cluster around amyloid plaques. This observation raises a question about the efficiency of APOE3-Jac variant to bind APOE receptors including microglial TREM2 and the ability to acquire the phenotype of disease-associated microglia (DAM).
A positive consequence of this study is that it may reinforce work on APOE biology leading to understanding mechanisms of AD risk and protection mediated by APOE.
References:
Tcw J, Qian L, Pipalia NH, Chao MJ, Liang SA, Shi Y, Jain BR, Bertelsen SE, Kapoor M, Marcora E, Sikora E, Andrews EJ, Martini AC, Karch CM, Head E, Holtzman DM, Zhang B, Wang M, Maxfield FR, Poon WW, Goate AM. Cholesterol and matrisome pathways dysregulated in astrocytes and microglia. Cell. 2022 Jun 23;185(13):2213-2233.e25. PubMed. BioRxiv.
Lee SI, Jeong W, Lim H, Cho S, Lee H, Jang Y, Cho J, Bae S, Lin YT, Tsai LH, Moon DW, Seo J. APOE4-carrying human astrocytes oversupply cholesterol to promote neuronal lipid raft expansion and Aβ generation. Stem Cell Reports. 2021 Sep 14;16(9):2128-2137. Epub 2021 Aug 26 PubMed.
Hong Kong University of Science & Technology
This study reports a rare APOE coding mutation, APOE3-Jacksonville (APOE3-Jac; or rs199768005), which is located adjacent to the ApoE lipid-binding regions and exerts AD-protective effects by reducing the brain Aβ load in humans. Meanwhile, restricting the expression of the ApoE3-Jac protein in astrocytes leads to a reduction of the brain Aβ load in amyloidosis transgenic 5XFAD mice. The authors also demonstrate the roles of APOE3-Jac in modifying the biochemical properties of ApoE, including reducing the self-aggregation of ApoE and enhancing its lipid-binding properties. These data provide compelling evidence on the protective roles of this APOE mutation against AD pathogenesis.
The discovery of the common coding genetic variants, APOE-ε4 and APOE-ε2, that strongly associate with AD across different ethnic groups makes APOE a well-accepted genetic risk factor (Zhou, Fu, and Ip, 2021). The recent identification of rare coding AD-protective APOE mutations, such as the APOE3-Jac and Christchurch mutations, in European-descent populations further support the critical roles of APOE in AD pathogenesis (Arboleda-Velasquez et al., 2019).
Meanwhile, recent studies have also revealed the contribution of noncoding genetic risk factors in APOE and its surrounding regions to AD risk (Rajabli et al., 2018; Babenko et al., 2018). In particular, the noncoding variants and haplotypes in APOE-surrounding regions recently identified can modify the expression of APOE, exerting AD risk independent of APOE-ε4 (Zhou et al., 2019).
Notably, these noncoding variants, like APOE-ε4, also exhibit varied frequencies across different ethnic groups (Zhou et al., 2019; Maestre et al., 1995). Thus, it would be of interest to conduct a comprehensive analysis for common and rare coding risks in APOE and nearby regions in non-European descent populations. In particular, characterizing the biological properties of APOE rare coding mutations would facilitate further understanding of the roles of APOE and its associated pathways in AD, as well as provide hints for the development of AD intervention strategies targeting APOE.
References:
Arboleda-Velasquez JF, Lopera F, O'Hare M, Delgado-Tirado S, Marino C, Chmielewska N, Saez-Torres KL, Amarnani D, Schultz AP, Sperling RA, Leyton-Cifuentes D, Chen K, Baena A, Aguillon D, Rios-Romenets S, Giraldo M, Guzmán-Vélez E, Norton DJ, Pardilla-Delgado E, Artola A, Sanchez JS, Acosta-Uribe J, Lalli M, Kosik KS, Huentelman MJ, Zetterberg H, Blennow K, Reiman RA, Luo J, Chen Y, Thiyyagura P, Su Y, Jun GR, Naymik M, Gai X, Bootwalla M, Ji J, Shen L, Miller JB, Kim LA, Tariot PN, Johnson KA, Reiman EM, Quiroz YT. Resistance to autosomal dominant Alzheimer's disease in an APOE3 Christchurch homozygote: a case report. Nat Med. 2019 Nov;25(11):1680-1683. Epub 2019 Nov 4 PubMed.
Babenko VN, Afonnikov DA, Ignatieva EV, Klimov AV, Gusev FE, Rogaev EI. Haplotype analysis of APOE intragenic SNPs. BMC Neurosci. 2018 Apr 19;19(Suppl 1):16. PubMed.
Maestre G, Ottman R, Stern Y, Gurland B, Chun M, Tang MX, Shelanski M, Tycko B, Mayeux R. Apolipoprotein E and Alzheimer's disease: ethnic variation in genotypic risks. Ann Neurol. 1995 Feb;37(2):254-9. PubMed.
Rajabli F, Feliciano BE, Celis K, Hamilton-Nelson KL, Whitehead PL, Adams LD, Bussies PL, Manrique CP, Rodriguez A, Rodriguez V, Starks T, Byfield GE, Sierra Lopez CB, McCauley JL, Acosta H, Chinea A, Kunkle BW, Reitz C, Farrer LA, Schellenberg GD, Vardarajan BN, Vance JM, Cuccaro ML, Martin ER, Haines JL, Byrd GS, Beecham GW, Pericak-Vance MA. Ancestral origin of ApoE ε4 Alzheimer disease risk in Puerto Rican and African American populations. PLoS Genet. 2018 Dec;14(12):e1007791. Epub 2018 Dec 5 PubMed.
Zhou X, Chen Y, Mok KY, Kwok TC, Mok VC, Guo Q, Ip FC, Chen Y, Mullapudi N, Alzheimer’s Disease Neuroimaging Initiative, Giusti-Rodríguez P, Sullivan PF, Hardy J, Fu AK, Li Y, Ip NY. Non-coding variability at the APOE locus contributes to the Alzheimer's risk. Nat Commun. 2019 Jul 25;10(1):3310. PubMed.
Zhou X, Fu AK, Ip NY. APOE signaling in neurodegenerative diseases: an integrative approach targeting APOE coding and noncoding variants for disease intervention. Curr Opin Neurobiol. 2021 Aug;69:58-67. Epub 2021 Feb 26 PubMed.
Macquarie University
This is an impressive piece of work focusing on the possible mechanism of the protective effect of the APOE3-Jac variant. The evidence provided is very convincing that this variant reduces APOE aggregation and Aβ pathology even with APOE4.
In my opinion, it provides a promising lead for a therapeutic approach. However, as the authors point out, they did not see an effect on cognition. This may lead to a similar controversy as Aduhelm has caused, i.e., great reduction in amyloid reduction but questionable clinical benefits.
These studies definitely need to be extended to an animal model that includes tau pathology, as the 5xFAD used here is a limitation.
Make a Comment
To make a comment you must login or register.