Excitotoxicity: Could a New Class of Drug Stop a Bad TRP?
Quick Links
For decades, the duality of NMDA glutamate receptors has perplexed researchers. Essential players in synaptic activity, plasticity, and learning, these receptors can also trigger a toxic intraneuronal cascade that decimates neurons in diseases such as Alzheimer’s, Huntington’s, amyotrophic lateral sclerosis, and stroke. How can that be? Now, new findings solve the riddle—and perhaps identify a new drug target. In today’s Science, researchers led by Hilmar Bading, University of Heidelberg, Germany, report that NMDARs bind to a specific type of cation channel. Blocking this interaction with a peptide decoy, or small molecules, prevented excitotoxic death in cultured neurons and in mouse models of stroke and retinal damage. The work establishes a new line of NMDA receptor research and, potentially, a new class of drug for neurodegeneration.
- Extrasynaptic NMDA receptors cause excitotoxicity in neurodegenerative diseases.
- They form complexes with TRPM4 cation channels.
- Blocking this interaction protects mice from stroke.
“This is a fascinating and elegant study, opening up a new potential therapeutic pathway to treat neurologic diseases that manifest extrasynaptic NMDAR hyperactivity, including AD,” wrote Stuart Lipton, The Scripps Research Institute, La Jolla, California (see full comment below). Lipton and others had previously linked Aβ and other misfolded proteins to NMDA receptor excitotoxicity (May 2011 news; June 2013 news).
Reconciling the synaptic and extrasynaptic effects of NMDA receptors has proven challenging. Some proposed that subunit compositions explained the dichotomy, but Bading told Alzforum he never really believed that. “I thought that the effects were subunit-independent and that the spatial aspect was the key,” he said. In 2017, he proposed that an unknown NMDAR-interacting protein completed an extrasynaptic complex that touches off a deadly triad of mitochondrial dysfunction, transcriptional deregulation, and structural disintegration, aka excitotoxicity (see image below and Bading, 2017). No one had identified this mysterious protein—until now.
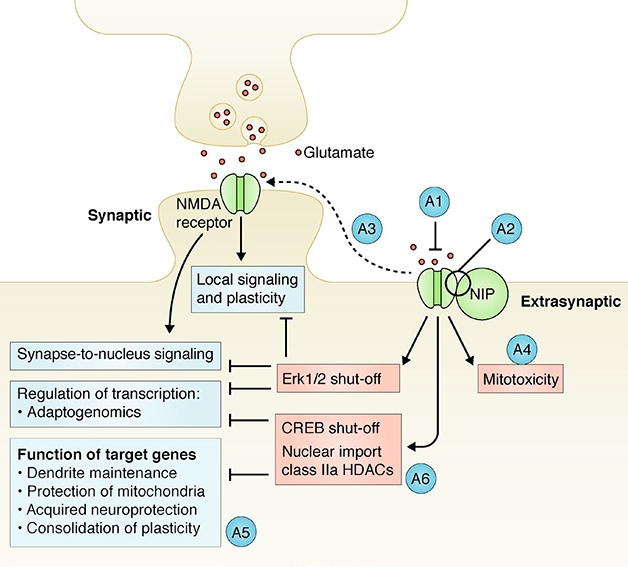
NIP in the Bud? Previously, Hilmar Bading predicted that an NMDA receptor-interacting protein (NIP) could explain why extrasynaptic NMDARs unleash a toxic signaling cascade (peach), while synaptic receptors are beneficial (blue). A1-A6 denote potential points for therapeutic intervention. [Courtesy of Bading, J Exp Med 2017.]
First author Jing Yan and colleagues went after some prime suspects. They focused on members of the transient receptor potential (TRP) channel family. TRPs funnel cations, though not calcium, through cell membranes, and they have been linked to neurodegeneration (Schattling et al., 2012; Sun et al., 2009; Aarts et al., 2003). Yan found that knocking down TRP sub-family M, member M4 (TRPM4), protected primary hippocampal neurons against glutamate, but not against the type of cell death that happens when scientists withdraw growth factors, suggesting the channel had some specificity for causing excitotoxicity.
Immunoprecipitation experiments confirmed that TRPM4 bound the two NMDAR subunits GluN2A and GluN2B, both in cultured neurons and in mouse brain. Next, Yan identified a 57 amino acid stretch in TRPM4, called TwinF, as the glue that held the complex together. TwinF, named for two phenylalanines at positions 666 and 667 of TRPM4, all but eliminated glutamate excitotoxicity when used as a decoy in hippocampal neurons. Equivalent decoys derived from a homologous section of TRPM5, the closest relative of TRPM4, had no effect, suggesting that only TRPM4 forms a death receptor complex with NMDARs.

New Concept–New Therapy? The NMDAR-TRPM4 (N/T) complex triggers depolarization of mitochondria and cell death (left). A computational screen (center) predicted that two small molecules would bind the N/T interface (center). These prevented excitotoxicity (right). [Courtesy of Yan et al., 2020 Science/AAAS.]
Yan found that TwinF bound to a stretch of 18 amino acids on the cytosolic side of GluN2A and GluN2B, close to the membrane. Bading dubbed this region I4 because it contains four regularly spaced isoleucines. Other glutamate receptor subunits lack this motif.
The researchers went beyond identifying this complex. Using computational analysis, they screened more than 1 million compounds to find two that might disrupt the NMDAR/TRPM4 interface. Compounds 8 and 19 were predicted to bind different locations near the TwinF motif of TRPM4 (see image below).
These compounds prevented mitochondrial dysfunction and cell death in hippocampal neuron cultures. They also increased synaptic NMDAR signaling, since they increased phospho-CREB, p-ERK, and transcription of the immediate/early gene c-fos, all without affecting calcium signals. In contrast, while NMDAR blockers, such as MK-801 and DL-APV, can weaken excitotoxicity and protect neurons, they also suppress synaptic NMDAR signaling. Bading and colleagues believe that compounds 8 and 19 in effect detoxify NMDAR signaling.
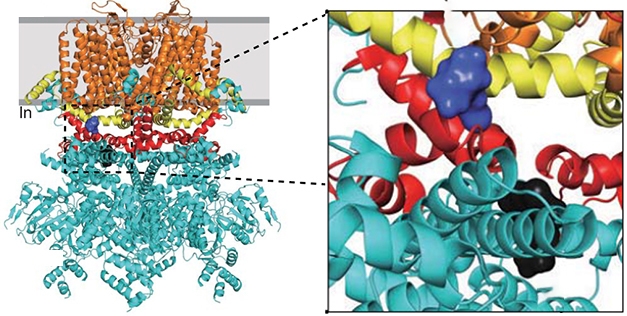
Butting In: This model of the TRPM4 channel shows where compound 8 (purple) and compound 19 (black) bind to the TwinF motif (red), which lies between the cytoplasmic domain (cyan), TRP (yellow), and transmembrane domains (orange). [Courtesy of Yan et al., 2020 Science/AAAS.]
Compound 8 protected neurons in mouse models of stroke and retinal degeneration. Five intraperitoneal injections of it, beginning 16 hours before a middle cerebral artery occlusion, with the last given 12 hours after, reduced the infarct area in the brain seen a week later. The same regimen reduced retinal cell degeneration one week after the researchers injected NMDA into mouse vitreous, the gel-like fluid inside the eyes (see image below).
In a separate experiment, the researchers tracked NMDAR/TRPM4 complexes in cortical lysates using immunoprecipitation. Two to six hours after intraperitoneal injection of compound 8, the amount of these brain complexes the scientists were able to isolate fell by up to 38 percent. A day later, complex levels had returned to normal, implying the drug effect is reversible. The scientists did not test compound 19 in vivo, because it caused cell death at high concentrations.

Eyeball This. Immunohistochemistry of the retinal ganglion cell marker Brn3a reveals loss of cells after intravitreal injection of NMDA (right panels). Intraperitoneal injection of compound 8 (bottom panels) protected the neurons. [Courtesy of Yan et al., 2020, Science/AAAS.]
All told, the data indicate the scientists have discovered a new type of NMDAR complex that explains glutamate excitotoxicity, and potentially two new classes of drugs to prevent it. “Both compounds are amazing,” Bading said. “Compound 8 is no bigger than aspirin or ibuprofen. It looks harmless, disrupts the complex, and eliminates toxicity.” Based on the structure, Bading thinks it could be given by mouth and probably crosses the blood-brain barrier. The success of the intraperitoneal treatment in reducing infarct in the brain supports this idea.
Bading is collaborating with other groups to test the molecules in other disease models, including those for AD and ALS. FundaMental Pharma, a biotech company he started some years ago, holds patents on these compounds. Bading is looking for partners to help with drug development.
In the meantime, Bading acknowledged that fundamental questions remain. How does the NMDAR/TRPM4 complex cause neurodegeneration? Not via normal NMDAR or TRPM4 activity, since neither TwinF nor compound 8 block these functions. “We know the death complex works, but we don’t know how it works,” Bading said. “My hunch is it damages mitochondria. This is speculation, but it could be that the NMDAR/TRPM4 complex lies near mitochondria, or maybe mitochondria are even being recruited via another protein in some sort of tripartite complex,” he said.
He believes calcium may start the death spiral. “We know that the global calcium signal is unchanged, so the idea of calcium overload is out the window, but a high localized calcium increase could initiate the deterioration process.”
As to whether NMDARs and TRPM4 bind each other purely by chance, or because the complex fulfills a physiological role, again Bading was tempted to speculate. He proposed that TRPM4 holds NMDARs in the membrane as a type of reservoir that could be called upon when neurons are trying to remodel synapses. “Lateral diffusion is a beautiful mechanism for remodeling of synapses,” he said, “because it would be faster and consume less energy than constant endo- and exocytosis.” In the healthy brain, the complex would pose no problem, because neurons and glia efficiently scoop glutamate out of the extracellular space. However, when a toxin, for example Aβ in the case of AD, impedes this, or when cells start pumping out glutamate, then things can start to go badly. “That’s when this potentially dangerous receptor is activated,” Bading said.—Tom Fagan
References
News Citations
Paper Citations
- Bading H. Therapeutic targeting of the pathological triad of extrasynaptic NMDA receptor signaling in neurodegenerations. J Exp Med. 2017 Mar 6;214(3):569-578. Epub 2017 Feb 16 PubMed.
- Schattling B, Steinbach K, Thies E, Kruse M, Menigoz A, Ufer F, Flockerzi V, Brück W, Pongs O, Vennekens R, Kneussel M, Freichel M, Merkler D, Friese MA. TRPM4 cation channel mediates axonal and neuronal degeneration in experimental autoimmune encephalomyelitis and multiple sclerosis. Nat Med. 2012 Dec;18(12):1805-11. Epub 2012 Nov 18 PubMed.
- Sun HS, Jackson MF, Martin LJ, Jansen K, Teves L, Cui H, Kiyonaka S, Mori Y, Jones M, Forder JP, Golde TE, Orser BA, Macdonald JF, Tymianski M. Suppression of hippocampal TRPM7 protein prevents delayed neuronal death in brain ischemia. Nat Neurosci. 2009 Oct;12(10):1300-7. Epub 2009 Sep 6 PubMed.
- Aarts M, Iihara K, Wei WL, Xiong ZG, Arundine M, Cerwinski W, Macdonald JF, Tymianski M. A key role for TRPM7 channels in anoxic neuronal death. Cell. 2003 Dec 26;115(7):863-77. PubMed.
Further Reading
Papers
- Xia P, Chen HS, Zhang D, Lipton SA. Memantine preferentially blocks extrasynaptic over synaptic NMDA receptor currents in hippocampal autapses. J Neurosci. 2010 Aug 18;30(33):11246-50. PubMed.
Primary Papers
- Yan J, Bengtson CP, Buchthal B, Hagenston AM, Bading H. Coupling of NMDA receptors and TRPM4 guides discovery of unconventional neuroprotectants. Science. 2020 Oct 9;370(6513) PubMed.
Annotate
To make an annotation you must Login or Register.
Comments
The Scripps Research Institute, and University of California, San Diego
This is a fascinating and elegant study, opening up a new potential therapeutic pathway to treat neurologic diseases that manifest extrasynaptic NMDAR hyperactivity—including AD. The Bading approach allows us to potentially intervene at a downstream event, TRPM4 channel activity, with important therapeutic implications. I think it is a fantastic study and hope to learn more about this pathway in the near future from Hilmar Bading and his colleagues.
University of Edinburgh
Downstream of dysfunctional astrocytes in AD, one process that loss of glutamate homeostasis may converge on is excitotoxic neuronal and synapse loss. Deficits in glutamate uptake capacity due to glutamate transporter hypo-expression, bioenergetic deficits associated with vascular impairments, and Aβ exposure to astrocytes can all lead to elevate ambient glutamate and activation of extrasynaptic NMDARs. Bading’s lab found years ago that these receptors seem particularly adept at coupling to pro-death pathways compared to synaptic ones, something recapitulated and built on in other disease models by the likes of Stuart Lipton, Lynn Raymond and Michael Kreutz. However, the molecular basis for this has eluded researchers till now. The uncoupling of NMDARs from TRPM4 has the potential for being the Holy Grail of allowing physiological NMDAR signaling while preventing the pathological consequences of extrasynaptic NMDAR activation. An exciting discovery, and we can hope that such approaches can influence chronic neurodegenerative disease trajectory.
Make a Comment
To make a comment you must login or register.