PET Tracer Detects Synapse Loss Across Alzheimer’s Brain
Quick Links
Part 1 of a 2-part story.
A PET tracer that lights up synapses is able to detect a loss of connectivity across the Alzheimer’s brain, according to new research presented at the Human Amyloid Imaging conference, held January 15–17 in Miami, Florida. This new evidence of widespread loss in cortical regions broadly extends previous hints that the tracer, UCB-J, picks up synaptic loss in the hippocampi of AD patients. It holds out the prospect of live imaging of a phenomenon that neuropathologists have been describing for many years. “For the first time, we’re seeing with UCB-J PET in vivo what we expected to see based on the autopsy studies,” Christopher van Dyck of Yale University, New Haven, Connecticut, the senior author of the research, told Alzforum. In this study, lower UCB-J uptake also correlated with worse cognitive performance.
- As methods get better, PET tracer UCB-J detects synapse loss in AD cortex.
- UCB-J uptake correlates strongly with tangles, weakly with plaques.
- Synapse loss is associated with cognitive decline, as expected.
Other presentations at HAI linked decreased UCB-J signal to more plaques and tangles as seen by PET. Overall, the emerging data strengthen the case that this tracer could be used to help diagnose Alzheimer’s disease, and perhaps even detect a slowing of synapse loss with treatment, researchers said. However, so far, few people have been scanned, and scientists are repeating the findings in larger cohorts. They are also beginning to collect longitudinal data and scan people at presymptomatic disease stages to find out when synapses start to disappear.
Broadly speaking, studies increasingly combine imaging with multiple tracers, and these emerging data are helping scientists stage which biomarker changes follow others in the long process of Alzheimer’s pathogenesis. The same methods are shedding light on other neurodegenerative diseases. Some researchers in Miami reported decreased UCB-J uptake in frontotemporal dementia, Parkinson’s disease, and progressive supranuclear palsy, as well (see Part 2 of this series).
Developed by the Belgian pharma company UCB and first tested at Yale, UCB-J binds to the presynaptic vesicle protein SV2A (Jul 2016 news). In an initial study of 10 AD patients and 11 controls, van Dyck and colleagues found that patients took up less UCB-J than did controls in their hippocampus, though they were unable to see a difference in cortical regions (Dec 2017 conference news; Aug 2018 conference news).

More Amyloid, Fewer Synapses. In healthy people (left), the synaptic PET signal (top) is high, while amyloid PET (bottom) is low; in people with AD (right), the opposite is true. [Courtesy of Adam Mecca and Chris van Dyck.]
Dwindling Synaptic Signal Flags Alzheimer’s Disease
In Miami, Adam Mecca of Yale described how the methodology for analyzing the UCB-J signal had improved. Instead of using the centrum semiovale, a patch of white matter above the lateral ventricles, as the reference region, the researchers turned to the cerebellum. There, tracer uptake was less variable from person to person, lowering noise and sharpening the signal. Using this method, Mecca and colleagues scanned 19 cognitively normal controls, 14 people with mild cognitive impairment due to AD, and 20 with AD dementia.
In every brain region examined, amyloid-positive people bound less tracer than did controls (see image above, part A). The differences were biggest in the hippocampus and entorhinal cortex, but were statistically significant across the board. After correcting for brain atrophy, the signal remained significant in the hippocampus and amygdala, as well as in the entorhinal, lateral temporal, parietal, pericentral, and prefrontal cortex.
The uncorrected signal probably represents the total number of synapses in the region, while the corrected one indicates synaptic density in the remaining tissue; both are potentially interesting, van Dyck told Alzforum. Overall, the findings match the pattern of synaptic loss seen in postmortem AD brain.
And UCB-J binding mattered. More signal tracked with better scores on the CDR-SB and on a measure of episodic memory. This association between tracer uptake and cognition was robust across the whole cohort, though within the MCI and AD groups alone, it was weak. Possibly, synapse numbers vary enough between individuals to obscure disease effects in such small cohorts, van Dyck suggested. More sensitive cognitive measures may be needed to pick up the earliest effects of a modest loss in connectivity. The early studies that first described the close relationship between synaptic loss and cognitive decline were done in postmortem samples from people at advanced stages of dementia, van Dyck noted (e.g. Terry et al., 1991; DeKosky et al., 1996). Longitudinal data in larger cohorts will clarify the issue.
Other research at HAI hinted at a link between synapse loss and cognitive impairment in general. In Miami, Alexandra DiFilippo of the University of Wisconsin-Madison, presented preliminary data from UCB-J scans of participants recruited from the UW Alzheimer’s Disease Research Center. Working with Sterling Johnson, Barbara Bendlin, and Bradley Christian, DiFilippo has scanned 15 study participants so far with UCB-J, the tau tracer MK-6240, and PiB PET. Eleven of them were amyloid-negative controls, one was amyloid-positive but cognitively normal, and three were amyloid-negative but cognitively impaired. Compared with controls, the amyloid-positive person had a slight drop in UCB-J uptake in his or her hippocampus and inferior parietal and medial frontal lobes. The impaired participants had larger declines in hippocampus, parahippocampus, and entorhinal cortex. The synaptic losses in these cases of non-AD dementia suggest that this tracer could be useful for tracking many neurodegenerative conditions, DiFilippo noted.
The researchers are now scanning more participants, hoping to enroll 120. Ideally, 30 of them will have MCI, 30 AD dementia, DiFilippo said. All participants will be scanned at two time points. UW-Madison is partnering with the Yale group on synaptic imaging grants and harmonizing their methodology so they can pool data.
However, as usual in emerging AD research, things are not cut-and-dried. A recent autoradiography study of seven AD cases and seven controls reported no difference in UCB-J binding in postmortem samples (Metaxas et al., 2019). In Miami, some researchers noted that they, too, saw no large difference between AD and control UCB-J scans. However, others in Miami reported excellent results from quantitative autoradiography. The discrepancies may reflect methodological differences, such as choice of reference region.
“The past three years have been spent in method development and confirming that the tools we’ve been using do reflect the underlying biological reality,” noted Eugenii (Ilan) Rabiner of King’s College and Invicro, an imaging services company based in London. With better tools in hand, researchers can begin to ask broader questions about how synapse loss changes in aging and disease, Rabiner added.
Plaques, Tangles, Synapses: A Complicated Trio
How do changes in SV2A correlate with other markers of Alzheimer’s disease? Preliminary data suggest but a rough correspondence with plaque. At HAI, Ryan O’Dell in the Yale group reported that, among 19 controls, 14 people with MCI due to AD, and 24 with AD dementia, higher PiB PET signal correlated with lower UCB-J signal (see image above, part B). However, this relationship was driven by the difference between the AD groups and controls. Within the MCI due to AD and AD dementia groups alone, amyloid PET did not relate to synaptic PET. In fact, medial temporal cortex, the cortical region with the least fibrillar amyloid, had the greatest drop in UCB-J uptake compared with controls. Perhaps this is because only healthy neurons can make amyloid, and the profound neuron death in this region lowers both amyloid production and synapse number, van Dyck speculated.
Tangles appear to correspond more closely with synapses. Mecca compared flortaucipir to UCB-J uptake in 10 cognitively normal controls and 10 with AD; half of whom were at the MCI, half at the dementia stage. In all brain regions examined, people with AD had more flortaucipir uptake than controls, and in most of those regions, they also had fewer synapses. Again, the decrease in UCB-J was most profound in hippocampus and entorhinal cortex. Tangles in EC correlated with fewer synapses in the hippocampus, which would be expected if tauopathy attacked neurons that project from the EC to the hippocampus. In future work, the Yale group will switch from flortaucipir to Merck’s tau tracer MK-6240, in order to harmonize their data with the UW study.
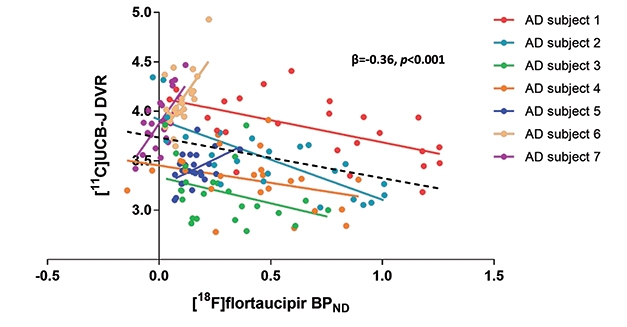
Stage-Specific Response? A spaghetti plot of seven participants shows a negative correlation between tangles and synapses in four people with high neocortical tau pathology, but positive in three people with low neocortical tau. [Courtesy of Bart van Berckel and Emma Coomans.]
Other groups are also correlating UCB-J with AD biomarkers. At HAI, Emma Coomans of Vrije University, Netherlands, showed data on UCB-J and flortaucipir scans in seven amyloid-positive AD patients. Working with Bart van Berckel, she found that within the group, higher flortaucipir uptake came with lower UCB-J uptake, similar to the results from Yale. However, when looking at individuals, the researchers saw a dichotomous response. In four people who had high flortaucipir signal in their neocortices, the more tangles they had in a given region, the lower the synaptic signal was in that region. The other three people had low neocortical tangles; in them, more tangles in a given brain region associated with a higher synaptic signal there (see image above).
Coomans suggested that this may be because high synaptic density in a region might, at first, actually attract and facilitate tangle pathology as it travels through synaptically connected circuits. On the other hand, it could reflect compensatory synaptic upregulation at early stages of disease, followed by synapse loss later. Some early studies have reported larger synapses in AD brain, as well as axonal sprouting and formation of new synapses (Scheff et al., 1990; Dekosky and Scheff, 1990; Cotman and Anderson, 1988). To see if their results hold up, the researchers will scan 30 people and follow them over time. They will also compare UCB-J imaging to magnetoencephalography, a different measure of synaptic activity.
Researchers are eager to start scanning people at presymptomatic stages. Synapse loss may be the earliest marker of degeneration, before atrophy, van Dyck believes. How early does it start? Can it be detected before cognitive decline manifests on tests? And if synapses vanish that early, might the UCB-J signal correlate better with amyloid accumulation at that stage than later on?
In cell culture, Aβ oligomers damage dendritic spines. Pedro Rosa-Neto, McGill University, Montreal, believes that amyloid may be the culprit behind synaptic loss at preclinical stages, while tangles may kill synapses later. “Amyloid is not necessarily associated with a change in cognition, but it is associated with a change in synaptic markers,” Rosa-Neto told Alzforum. Alternatively, van Dyck speculated that Aβ oligomers and pathological forms of tau might interact at early disease stages to harm synapses.—Madolyn Bowman Rogers
References
News Citations
- Multimodal Imaging of Neurodegenerative Diseases Links Pathology and Cellular Dysfunction
- Next Up for Human Brain Imaging: Synaptic Density?
- At CTAD, Tau PET Emerges as Favored Outcome Biomarker for Trials
- PET Ligand Lights Up AAIC, May Detect Synapse Loss in AD
Paper Citations
- Terry RD, Masliah E, Salmon DP, Butters N, DeTeresa R, Hill R, Hansen LA, Katzman R. Physical basis of cognitive alterations in Alzheimer's disease: synapse loss is the major correlate of cognitive impairment. Ann Neurol. 1991 Oct;30(4):572-80. PubMed.
- DeKosky ST, Scheff SW, Styren SD. Structural correlates of cognition in dementia: quantification and assessment of synapse change. Neurodegeneration. 1996 Dec;5(4):417-21. PubMed.
- Metaxas A, Thygesen C, Briting SR, Landau AM, Darvesh S, Finsen B. Increased Inflammation and Unchanged Density of Synaptic Vesicle Glycoprotein 2A (SV2A) in the Postmortem Frontal Cortex of Alzheimer's Disease Patients. Front Cell Neurosci. 2019;13:538. Epub 2019 Dec 5 PubMed.
- Scheff SW, DeKosky ST, Price DA. Quantitative assessment of cortical synaptic density in Alzheimer's disease. Neurobiol Aging. 1990 Jan-Feb;11(1):29-37. PubMed.
- Dekosky ST, Scheff SW. Synapse loss in frontal cortex biopsies in Alzheimer's disease: correlation with cognitive severity. Ann Neurol. 1990 May;27(5):457-64. PubMed.
- Cotman CW, Anderson KJ. Synaptic plasticity and functional stabilization in the hippocampal formation: possible role in Alzheimer's disease. Adv Neurol. 1988;47:313-35. PubMed.
Further Reading
Annotate
To make an annotation you must Login or Register.
Comments
No Available Comments
Make a Comment
To make a comment you must login or register.