Gently Used: Can Recycled Microglia Receptors Prevent Plaque?
Quick Links
Microglia do much of the heavy lifting when it comes to removing Aβ, but how do they do it? Researchers led by Douglas Green, St. Jude Children’s Research Hospital, Memphis, Tennessee, have identified an endocytic recycling process by which microglia clear β-amyloid and return used Aβ receptors back to the surface, the better to gobble more aggregated protein. In a paper published online June 27 in Cell, the scientists report that disrupting this process leads Aβ to build up outside microglia and clump into plaques.
- Microglia use LANDO, a form of endocytosis, to clear Aβ and recycle surface Aβ receptors.
- Without LANDO, Aβ builds up outside the cells and forms plaques.
- One AD mouse line lacking LANDO develops pathology and memory problems sooner.
The researchers call the process LANDO. That’s short for LC3-associated endocytosis, named for the LC3 protein associated with the Aβ-containing vesicles. In a mouse model of Alzheimer’s disease, disrupting LANDO brought on AD neuropathology or behavioral deficits earlier than is typical in these mice. The expression of proteins needed for LANDO wanes with age and AD, therefore a gradual failure of LANDO might play into the etiology of disease. “If we figure out how to improve LANDO, especially in aging, we think we’ve got a good chance to restore normal homeostasis and return to a healthier brain environment,” Green told Alzforum.
“The findings are quite striking,” said CongCong He, Northwestern University, Chicago. “How we maintain the number and function of Aβ receptors at the cell surface is an important question.”
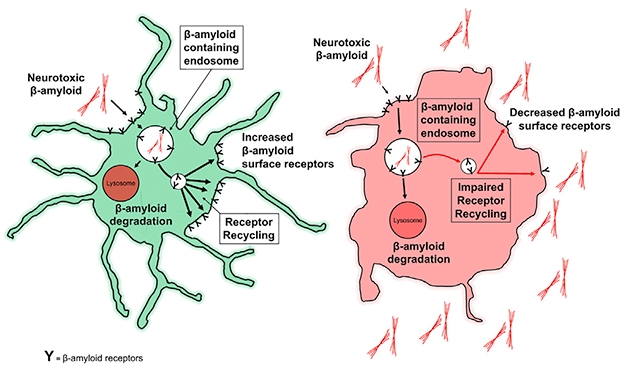
Rinse, Repeat? Aβ receptors on the surface of healthy microglia (green) bind β-amyloid and drag it off to be degraded through LANDO, and are then recycled back to the membrane. Without LANDO, recycling sputters, and β-amyloid builds up outside cells. [Courtesy of Heckmann et al., 2019. Cell.]
Green and colleagues initially set out to discover whether microglial autophagy played a role in clearance of Aβ. Autophagy, through which cells degrade unnecessary components, is known to be defective in neurodegenerative disorders, but has been studied mostly in neurons (for a review, see Harris and Rubinsztein, 2012). Could it also be responsible for the uptake of Aβ by microglia?
First author Bradlee Heckman and colleagues used 5XFAD mice and genetically deleted one of three autophagy genes from their microglia—the autophagy regulators FIP200 or ATG5, or the autophagy inhibitor Rubicon. Surprisingly, knocking out either ATG5 or Rubicon both led to a marked increase in Aβ deposition, though knocking out FIP200 did not. In addition, the ATG5 and Rubicon knockouts exhibited many AD-related phenotypes. Activated microglia released inflammatory cytokines and tau became hyperphosphorylated. In hippocampal neurons, reduced synaptic transmission led to impaired long-term potentiation and more neurons died. In addition, the mice did poorly at recognizing novel objects and exploring the Y-maze. All this happened when mice were just 4 months old, long before control 5XFAD mice showed pathology or behavioral deficits.
“This is dramatic at 4 months of age,” said Green. “We are seeing these accelerated and pronounced characteristics of neurodegenerative disease by manipulating only microglia.”
Since FIP200 was unimportant for Aβ clearance, the results suggested microglia relied on something other than run-of-the-mill autophagy, though the process did involve some overlapping proteins. What could it be? Both ATG5 and Rubicon are required for a pathway Green has studied for many years, called LC3-associated phagocytosis, or LAP (Heckmann et al., 2017). A form of phagocytosis, LAP involves LC3-labeled phagosomes that clear extracellular particles including apoptotic cells and pathogens.
Could LAP be the mechanism for clearing Aβ in microglia? At first, the answer looked to be yes, as the cytoplasmic LC3 protein coated the Aβ-containing vesicles. However, instead of co-localizing with phagocytic markers, LC3 co-localized with endosomal ones. Heckmann and colleagues hypothesized that they had stumbled on a new form of endocytosis that controls microglial clearance of Aβ, and called it LANDO.
A previous study had implicated two other autophagy proteins, Beclin1 and VPS34, in the recycling of Aβ receptors CD36 and TREM2 back to the microglial surface (Lucin et al., 2013). Could ATG5 and Rubicon be doing the same thing in LANDO? Sure enough, knocking out ATG5 or Rubicon led to less TREM2, CD36, and another putative Aβ receptor, TLR4, being recycled back to the microglial surface (see image below).

Back For More. The Aβ receptors TLR4, Trem2, CD36 (red) return to the surface of microglia (blue) in control cells and FIP200 knockouts after engulfing Aβ. However, without ATG5 or Rubicon, this recycling does not happen. [Courtesy of Heckmann et al., 2019. Cell.]
Overall, the results suggested to the authors that the LANDO pathway helps microglia clear Aβ and boosts the number of Aβ receptors at its surface. It also keeps these phagocytic microglia from becoming pro-inflammatory. ATG5, Beclin1 and other autophagy-related proteins have been reported to decrease with age and AD, suggesting their functional processes, including LANDO, falter as people get older (Pickford et al., 2008; Lipinski et al., 2010).
“If we find a way to recycle more Aβ receptors to the cell surface, we may be able to maximize the phagocytic function of microglia and improve the removal of toxic Aβ,” He told Alzforum. She wondered how Aβ receptors are eventually sorted into recycling endosomes, and how LC3 and other autophagy proteins functioned in the process.
“This study provides further evidence that autophagy and its related proteins are important for AD-related pathologies in glial cells as well as neurons,” said Per Nilsson, Karolinska Institutet, Stockholm. This means targeting LANDO or other autophagic processes could be tricky, he said, since a drug for one protein could have disparate effects on different cell types. Highly selective compounds might make it possible, however, he said. The data are robust, he added, though it will be necessary to demonstrate LANDO in different mouse models with less extreme pathology.
For his part, Ralph Nixon, New York University, asked how specific LANDO is for Aβ receptors and for microglia. “The promise of this is the possibility that there is some highly selective purpose for LANDO in microglia that would lend itself to being specifically targeted,” he said. If instead LANDO recycles many other types of receptor, or exists in other cell types such as neurons and astrocytes, that would limit its specificity and possible use as a drug target.—Gwyneth Dickey Zakaib
References
Research Models Citations
Paper Citations
- Harris H, Rubinsztein DC. Control of autophagy as a therapy for neurodegenerative disease. Nat Rev Neurol. 2012 Feb;8(2):108-17. PubMed.
- Heckmann BL, Boada-Romero E, Cunha LD, Magne J, Green DR. LC3-Associated Phagocytosis and Inflammation. J Mol Biol. 2017 Nov 24;429(23):3561-3576. Epub 2017 Aug 25 PubMed.
- Lucin KM, O'Brien CE, Bieri G, Czirr E, Mosher KI, Abbey RJ, Mastroeni DF, Rogers J, Spencer B, Masliah E, Wyss-Coray T. Microglial beclin 1 regulates retromer trafficking and phagocytosis and is impaired in Alzheimer's disease. Neuron. 2013 Sep 4;79(5):873-86. PubMed.
- Pickford F, Masliah E, Britschgi M, Lucin K, Narasimhan R, Jaeger PA, Small S, Spencer B, Rockenstein E, Levine B, Wyss-Coray T. The autophagy-related protein beclin 1 shows reduced expression in early Alzheimer disease and regulates amyloid beta accumulation in mice. J Clin Invest. 2008 Jun;118(6):2190-9. PubMed.
- Lipinski MM, Zheng B, Lu T, Yan Z, Py BF, Ng A, Xavier RJ, Li C, Yankner BA, Scherzer CR, Yuan J. Genome-wide analysis reveals mechanisms modulating autophagy in normal brain aging and in Alzheimer's disease. Proc Natl Acad Sci U S A. 2010 Aug 10;107(32):14164-9. PubMed.
Further Reading
Papers
- Fujikura M, Iwahara N, Hisahara S, Kawamata J, Matsumura A, Yokokawa K, Saito T, Manabe T, Matsushita T, Suzuki S, Shimohama S. CD14 and Toll-Like Receptor 4 Promote Fibrillar Aβ42 Uptake by Microglia Through A Clathrin-Mediated Pathway. J Alzheimers Dis. 2019;68(1):323-337. PubMed.
- Maday S. Mechanisms of neuronal homeostasis: Autophagy in the axon. Brain Res. 2016 Mar 30; PubMed.
Primary Papers
- Heckmann BL, Teubner BJ, Tummers B, Boada-Romero E, Harris L, Yang M, Guy CS, Zakharenko SS, Green DR. LC3-Associated Endocytosis Facilitates β-Amyloid Clearance and Mitigates Neurodegeneration in Murine Alzheimer's Disease. Cell. 2019 Jul 25;178(3):536-551.e14. Epub 2019 Jun 27 PubMed. Correction.
Annotate
To make an annotation you must Login or Register.
Comments
No Available Comments
Make a Comment
To make a comment you must login or register.