TDP-43: Separating Physiological Wheat from Pathological Chaff
Quick Links
Up to now, toxic TDP-43 aggregates have been hard to isolate, making them difficult to characterize physically. A new technique from the lab of Magdalini Polymenidou, University of Zurich, may help. Called SarkoSpin, it separates the aggregates from TDP-43 oligomers bound to nucleic acids. Aggregates from patients with various subtypes of frontotemporal lobar degeneration (FTLD) have distinct densities, seeding activities, and toxicities. “This suggests that the differences we see at the clinical level in these FTLD subtypes might be encoded within the different conformations of the aggregates,” Polymenidou told Alzforum.
- The SarkoSpin method separates toxic TDP-43 from healthy oligomers.
- Aggregates from different subtypes of FTLD have distinct characteristics.
- The method paves the way for novel TDP-43 experiments and models.
“The authors have managed to separate the pathological form of TDP-43 from the physiological form,” said Manuela Neumann, German Center for Neurodegenerative Diseases, Tübingen, Germany, who was not involved in the work. “We needed such a protocol.”
Based on morphological features of TDP-43 inclusions and where they occur in the brain, researchers have defined several subtypes of FTLD that tend to have their own sets of clinical symptoms. Patients with FTLD-TDP type A (FTLD-A) develop neuronal cytoplasmic inclusions (NCIs) in upper cortical layers and typically show signs of behavioral-variant FTD. FTLD-B patients develop granular NCIs in all cortical layers and usually have either FTD or motor neuron disease. Those with FTLD-C have scattered inclusions in superficial cortical layers and develop semantic dementia. Are these inclusions made up of different strains of TDP-43 aggregates?
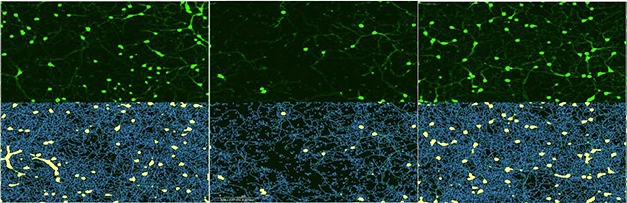
Varying toxicities. After 10 days, few neurons (green, top) survive added FTLD-TDP-A aggregates (middle), whereas neurons treated with FTLD-TDP-C (right) do as well as controls (left). Bottom panels show neuronal cell bodies (yellow) and neurites (blue). [Courtesy of Laferrière et al., 2018.]
This has proven difficult to study. Last year, for example, Polymenidou and colleagues reported that TDP-43 dimers form complexes with DNA and RNA that are resistant to solubilization by detergents such as sarkosyl, making it hard to physically separate these physiological species from pathological aggregates (Afroz et al., 2017).
“This signal-to-noise ratio has always been a limitation,” said Nicholas Seyfried, Emory University, Atlanta, who was not involved in the research. “If the picture is muddied by a mixture of pathological and physiological TDP-43, then it becomes challenging to resolve meaningful structural or biological differences.”
To solve this problem, first author Florent Laferrière and colleagues turned to nucleases. They added benzonase to tissue extracts to digest DNA and RNA, then the sarkosyl to solubilize the TDP-43 oligomers. After centrifugation, oligomers floated in the supernatant, while the insoluble aggregates spun down in the pellet.
Laferrière used this procedure on homogenized motor and frontal cortex tissue from 25 FTLD-A patients, three FTLD-B, 11 FTLD-C, 21 controls, and 13 people with amyotrophic lateral sclerosis (ALS), in which TDP-43 aggregates also develop. In controls, 99.9 percent of all TDP-43 wound up in the supernatant. In diseased brains, 12 percent of the protein ended up in the pellet. The pelleted protein bore all the hallmarks of pathological TDP-43, including polyubiquitination, C-terminal cleavage, and hyperphosphorylation.
However, the similarities ended there. When the researchers examined the protein content by mass spectrometry, they found different patterns in different pellets. For instance, on average, pellets from FTLD-A contained proteins such as ASHA1, OGDHL, and FBXO2. Those from FTLD-C had TXNL1, PRKCA, and CTTN. Those from ALS included PFN1, PLS3, and C8ORF46. There were too few samples in the FTLD-B group to average. There was no obvious commonality among these proteins, although some are known to be mutated or enriched in FTLD/ALS. Since none of these proteins aggregate or co-localize with TDP-43 in neurons, and none pelleted from control brain samples, the authors think that TDP-43 inclusions somehow rendered them insoluble. Others thought the appearance of unrelated proteins in the pellet was inconsistent, even within FTLD subtypes, and an indication that the procedure may still need refinement.
Still, the TDP-43 aggregates from the FTLD subtypes differed physically. Those from FTLD-C were denser than those from ALS or FTLD-A. Conformationally, the aggregates seemed different, too, since a monoclonal antibody that recognizes amino acids 203–209 of TDP-43 bound more weakly to FTLD-C material than FTLD-A, suggesting the epitope is hidden in the more densely packed aggregate.
Lastly, the authors found that the pellets had different seeding activity and toxicity. In cultured HEK293 cells that expressed a tagged form of TDP-43, aggregates from FTLD-A, but not –C, caused native protein to misfold and clump up. In primary cortical neurons from mice, aggregates from FTLD-A shrank neurites and killed cells within 10 days of exposure, while aggregates from FTLD-C and controls had no effect (see image above). The researchers have not yet analyzed the toxicity of pathological TDP-43 from ALS, Polymenidou said.
That pathological TDP-43 from different subtypes of FTLD have different molecular characteristics fits with the idea that there are different strains of TDP-43 aggregates, much like the prion-like strains proposed for α-synuclein, Aβ, and tau, wrote the authors (Jun 2015 news; Meyer-Luehmann et al., 2006; May 2014 news). Others felt the aggregates were not well-characterized. Ian Mackenzie, University of British Columbia, said that it would be useful to employ other biophysical techniques, such as mass spectrometry and cryoEM, to explore these isolates in detail.
Rosa Rademakers, Mayo Clinic, Jacksonville, Florida, said the work further validates the idea that different types of TDP-43 pathology occur in FTLD, but she wondered how the protein subtypes relate to genetic variants of the disease. Mutations in genes for progranulin, C9ORF72, tau, VCP, and TBK1 all cause FTLD. Rademakers noted that in this analysis, FTLD-A samples, for example, came from people with PGRN, C9ORF72, and sporadic FTLD. “In future analyses, characterizing TDP-43 from patients with known genetic mutations might tease out variability caused by the different genes,” she said. While the findings did not make it into the paper, Polymenidou said that they found no differences among FTLD-TDP-A aggregates from people with different familial mutations. “However, we cannot exclude that there are differences that are undetectable by our current methods. The largest differences that we can detect best correlate with the neuropathological type and not the genetic mutations.”
Polymenidou plans to test the seeding propensity of these aggregates in mice to see if they cause subtype-specific disease. “That needs to be done before we can call them strains,” she told Alzforum. She also wants to understand what gives these aggregates their different biochemical properties.
Recent research from the lab of Virginia Lee at the University of Pennsylvania, Philadelphia, suggests that human TDP-43 aggregates do indeed seed pathology in mice (Oct 2018 news). “Combining SarkoSpin with subsequent injection of isolated TDP-43 assemblies into mice could enable measurement of subtype-specific spreading of toxic TDP-43 assemblies in vivo,” wrote Edward Barbieri and James Shorter at the University of Pennsylvania in an accompanying News and Views. Such experiments could yield robust new models of ALS and FTLD subtypes, they wrote.—Gwyneth Dickey Zakaib
References
News Citations
- Shape of α-Synuclein Aggregates Influences Pathology
- Like Prions, Tau Strains Are True to Form
- TDP-43 Joins Cell-To-Cell Propagation Gang
Paper Citations
- Afroz T, Hock EM, Ernst P, Foglieni C, Jambeau M, Gilhespy LA, Laferriere F, Maniecka Z, Plückthun A, Mittl P, Paganetti P, Allain FH, Polymenidou M. Functional and dynamic polymerization of the ALS-linked protein TDP-43 antagonizes its pathologic aggregation. Nat Commun. 2017 Jun 29;8(1):45. PubMed.
- Meyer-Luehmann M, Coomaraswamy J, Bolmont T, Kaeser S, Schaefer C, Kilger E, Neuenschwander A, Abramowski D, Frey P, Jaton AL, Vigouret JM, Paganetti P, Walsh DM, Mathews PM, Ghiso J, Staufenbiel M, Walker LC, Jucker M. Exogenous induction of cerebral beta-amyloidogenesis is governed by agent and host. Science. 2006 Sep 22;313(5794):1781-4. PubMed.
Further Reading
Papers
- Mackenzie IR, Neumann M. Reappraisal of TDP-43 pathology in FTLD-U subtypes. Acta Neuropathol. 2017 Jul;134(1):79-96. Epub 2017 May 2 PubMed.
- Neumann M, Mackenzie IR. Review: Neuropathology of non-tau frontotemporal lobar degeneration. Neuropathol Appl Neurobiol. 2019 Feb;45(1):19-40. PubMed.
Primary Papers
- Laferrière F, Maniecka Z, Pérez-Berlanga M, Hruska-Plochan M, Gilhespy L, Hock EM, Wagner U, Afroz T, Boersema PJ, Barmettler G, Foti SC, Asi YT, Isaacs AM, Al-Amoudi A, Lewis A, Stahlberg H, Ravits J, De Giorgi F, Ichas F, Bezard E, Picotti P, Lashley T, Polymenidou M. TDP-43 extracted from frontotemporal lobar degeneration subject brains displays distinct aggregate assemblies and neurotoxic effects reflecting disease progression rates. Nat Neurosci. 2019 Jan;22(1):65-77. Epub 2018 Dec 17 PubMed.
- Barbieri EM, Shorter J. TDP-43 shapeshifts to encipher FTD severity. Nat Neurosci. 2019 Jan;22(1):3-5. PubMed.
Annotate
To make an annotation you must Login or Register.
Comments
University College London
University of Reading
It is well known that amyloidogenic proteins (i.e., proteins that tend to misfold and then aggregate in insoluble deposits during disease) have a tendency to behave in a prion-like fashion. In other words, they function as seeds for the misfolding of protein counterparts leading to protein aggregation. In the current work, the development of a novel method to isolate and purify proteins that constitute aggregates contributing to TDP-43 proteinopathy is evidenced. The strength of this work lies in that, in combination with mass-spectrometry, the authors have started shedding light on the molecular differences across TDP pathological subtypes, specifically, FTLD-TDP-A and FTLD-TDP-C. The significance of this work is that these features could represent a means to discriminate across FTLD subtypes molecularly and that a molecular definition of the pathogenic process might indicate impacted sub-cellular (and cell-specific) pathways that lead to disease, opening a concrete opportunity to identify molecular targets that are specific to the different subtypes of the TDP-FTD spectrum.
Here the authors are able to describe the microenvironment around and within the pathological lesions at a scale of resolution that has not been reached before. And they have found consistent and robust patterns that allow them to link protein pathology to a specific clinical phenotype (i.e. FTLD-TDP A and C).
The contribution of this manuscript is twofold. First, we get to know a bit more about the molecular composition of subcellular lesions caused by or in association with TDP-43 pathology, as well as their associated cell-specificity. For example, it is noteworthy that aggregate elements such as FBXO2 locate to astrocytes and not to neurons. This study has shown the relationship (physical and functional) between TDP-43 and FBXO2 is possibly more complex than previously thought, but at the same time, may help better interpret it. Second, these adjunct proteins might prove to be novel potential genetic candidates if they harbor deleterious variants that act as disease modifiers or determine the specific pathological subtype affecting and impacting FTLD and ALS cases (with TDP-43 pathology). At the very least, they represent a stepping stone in deciphering differential cellular vulnerability and differential pathogenic mechanisms.
Clearly, and as the authors stated, it will need to be verified if the seeding capacity and strain propagation are conserved in vivo, since this is not a matter the current work is able to answer. As well, the conundrum of cause versus consequence remains open for debate: is the specific clinical manifestation the driving force leading to different aggregated protein species (we already know from in vitro studies that amyloid proteins tend to form different types of aggregates based on the chemico-physical and temporal features of the experimental conditions)? Or is it the other way around, i.e., is it the propensity of TDP-43 to aggregate as type A or type C that drives and differentiates the clinical phenotypes? In other words, are the aggregates a consequence of, or the determinant of, the clinical subtype?
Make a Comment
To make a comment you must login or register.