With Tau in Synapses, NO Neurovascular Coupling
Quick Links
When too much tau invades dendrites, synaptic signaling goes haywire. Now, researchers led by Costantino Iadecola at Weill Cornell Medical College, New York, accuse this mislocalized tau of disrupting neurovascular coupling as well. In the August 10 Nature Neuroscience, they report that young mice overexpressing mutant human tau cannot amplify blood flow in their brains in response to neuronal activity. The defect was caused by soluble tau in the postsynapse. The protein displaced neuronal nitric oxide synthase, preventing synaptic activity from spurring nitric oxide production. Without NO, blood vessels did not expand. “This may be one of the earliest manifestations of tau pathology,” Iadecola told Alzforum. He believes this unresponsive vasculature could make it more difficult for the brain to clear toxic proteins, leading to further harmful effects.
- In mouse models of tauopathy, neurovascular coupling flags before tangles form.
- Soluble tau in the synapse displaces nitric oxide synthase.
- Without NO, blood vessels cannot dilate in response to neuronal activity.
“This work is exciting, helping define a possible route of tau-mediated neurodegeneration, and adding to a growing stack of evidence that tau and vasculature are intertwined,” Rachel Bennett at Massachusetts General Hospital, Boston, wrote to Alzforum (full comment below).
According to data from the Alzheimer’s Disease Neuroimaging Initiative, neurovascular dysfunction may be one of the earliest signs of AD, preceding even amyloid buildup (Jul 2016 news). Cardiovascular risk factors and amyloid burden together conspire to worsen tau pathology (Rabin et al., 2019). However, few studies have investigated the effects of tau pathology itself on the vasculature.
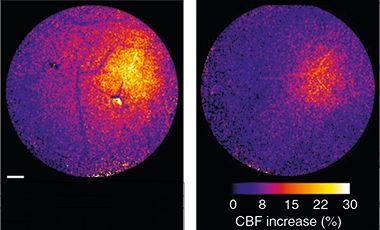
Muted. The activity-dependent increase in blood flow (color scale) is much greater in control (left) than PS19 tauopathy mice (right). [Courtesy of Park et al., Nature Neuroscience.]
First author Laibaik Park addressed this by examining cerebral blood flow in two different tauopathy mouse models, PS19 and rTg4510. Both express mutant human tau—P301S and P301L, respectively—and develop tangles after four to six months. Memory problems and neuron loss follow. In the rTg4510 line, the tau transgene is under the control of a tetracycline response element, and can be shut off by feeding the mice doxycycline.
The researchers assessed cortical blood flow through a cranial window at 2 to 3 months of age, before tangles formed. Blood moved at normal volume, and increased in response to agonists that act on endothelial and smooth muscle cells. However, the animals’ vasculatures had a specific defect in neurovascular coupling. When the researchers tweaked the mouse’s whiskers, the surge in blood flow in the barrel cortex was only half that in control mice (see image above).
Though these young mice had no tangles, tau was abnormally phosphorylated at Ser202/Thr205, as seen by immunostaining with the AT8 antibody, and this p-tau had accumulated in dendrites. Dendritic tau has been associated with Aβ-mediated excitotoxicity and with reduced synaptic signaling (Jul 2010 conference news; Sep 2010 news; Jan 2011 news). To find out if abnormal tau was responsible for the defects in neurovascular coupling, the authors turned off transgene production in the rTg4510 mice by feeding them doxycycline.
When tau expression was suppressed starting at 3 months of age, neurovascular coupling was normal four months later, suggesting that removing dendritic tau reverses defects. These mice also maintained cortical thickness and memory comparable to controls at 7 months, in contrast to deficits seen in rTg4510 mice with constant transgene expression. Strengthening the idea that the defects in neurovascular coupling were caused by soluble tau, the authors were able to reproduce them by adding recombinant wild-type or mutant tau directly to the barrel cortices of wild-type mice.
Why didn’t blood vessels respond in the presence of tau? Normally, neuronal activity triggers neuronal nitric oxide synthase (nNOS), which is bound to NMDA receptors in the postsynapse through the adaptor protein PSD95 (Kornau et al., 1995; Brenman et al., 1996). Activation of nNOS produces NO, which dilates blood vessels. In the tauopathy mouse models, however, less than half the normal amount of nNOS was present at the synapse, as seen by co-immunoprecipitation with PSD95. In addition, neurons isolated from these mice did not produce NO in response to NMDA, confirming a defect.
To explore the mechanism, the researchers expressed mutant P301L tau together with PSD95 and nNOS in cultured HEK293T cells, then used co-immunoprecipitations to see if any bound together. Tau and nNOS bound to the same site on PSD95. The findings imply that excess tau in dendrites displaces nNOS, disrupting signaling.
“These results directly link postsynaptic activity to cerebrovascular dysfunction, providing a novel concept for how tau abnormalities can lead to some of the earliest pathophysiological changes during AD progression,” Dezhi Liao at the University of Minnesota, Minneapolis, wrote to Alzforum (full comment below). He noted that the findings contrast with the prevailing belief that cerebral amyloid angiopathy is responsible for vascular problems in AD (Aug 2002 news). He suggested that future work might pin down which forms of tau are toxic by exploring the effects of physiological levels of wild-type and unphosphorylated human tau. All tau used in Iadecola’s experiments was phosphorylated at Ser202/Thr205.
What might loss of NO do to the brain? Iadecola noted that this signaling molecule does more than dilate blood vessels. It also stimulates neurogenesis in response to activity and it suppresses tau phosphorylation (Shen et al., 2019; Oct 2019 news). In addition, the NO-dependent change in blood vessel diameter has been linked to clearance of solutes such as Aβ from the brain (van Veluw et al., 2020). If tau interferes with NO production, that could lead to multiple defects that could worsen tau and even Aβ pathology, Iadecola suggested.
Park and colleagues are developing peptides that interfere with tau binding to PSD95 without harming nNOS binding. Once they have a good candidate, they will test it in mouse models to see if it preserves neurovascular coupling, Iadecola said. In theory, such a treatment could help people with AD, because Aβ causes tau to move into dendrites, as well as people with frontotemporal dementia, since tau mutations are common in that disorder.
Bennett noted that research presented at the virtual AD/PD conference in April suggests tau accumulation in human brain diminishes blood flow (see comment below). “I look forward to seeing this area of science continue to grow, and eagerly anticipate work from neuroimaging groups using combined PET/MRI techniques to assess similar relationships between tau and vasculature in human disease,” Bennett wrote. Intriguingly, some prior research has linked cerebrovascular disease to increased tangles and cognitive decline (May 2018 news).—Madolyn Bowman Rogers
References
News Citations
- LOAD of Data Place Vascular Malfunction as Earliest Event in Alzheimer’s
- Honolulu: The Missing Link? Tau Mediates Aβ Toxicity at Synapse
- The Plot Thickens: The Complicated Relationship of Tau and Aβ
- Tau’s Synaptic Hats: Regulating Activity, Disrupting Communication
- Amyloid in Transgenic AβPP Mice Affects Blood Flow
- Not Just Blood Pressure—Dietary Salt Linked to Tau Phosphorylation
- Cerebrovascular Disease: Does Tau Mediate Cognitive Decline?
Research Models Citations
Paper Citations
- Rabin JS, Yang HS, Schultz AP, Hanseeuw BJ, Hedden T, Viswanathan A, Gatchel JR, Marshall GA, Kilpatrick E, Klein H, Rao V, Buckley RF, Yau WW, Kirn DR, Rentz DM, Johnson KA, Sperling RA, Chhatwal JP. Vascular Risk and β-Amyloid Are Synergistically Associated with Cortical Tau. Ann Neurol. 2019 Feb;85(2):272-279. Epub 2019 Jan 7 PubMed.
- Kornau HC, Schenker LT, Kennedy MB, Seeburg PH. Domain interaction between NMDA receptor subunits and the postsynaptic density protein PSD-95. Science. 1995 Sep 22;269(5231):1737-40. PubMed.
- Brenman JE, Chao DS, Gee SH, McGee AW, Craven SE, Santillano DR, Wu Z, Huang F, Xia H, Peters MF, Froehner SC, Bredt DS. Interaction of nitric oxide synthase with the postsynaptic density protein PSD-95 and alpha1-syntrophin mediated by PDZ domains. Cell. 1996 Mar 8;84(5):757-67. PubMed.
- Shen J, Wang D, Wang X, Gupta S, Ayloo B, Wu S, Prasad P, Xiong Q, Xia J, Ge S. Neurovascular Coupling in the Dentate Gyrus Regulates Adult Hippocampal Neurogenesis. Neuron. 2019 Sep 4;103(5):878-890.e3. Epub 2019 Jun 27 PubMed.
- van Veluw SJ, Hou SS, Calvo-Rodriguez M, Arbel-Ornath M, Snyder AC, Frosch MP, Greenberg SM, Bacskai BJ. Vasomotion as a Driving Force for Paravascular Clearance in the Awake Mouse Brain. Neuron. 2020 Feb 5;105(3):549-561.e5. Epub 2019 Dec 3 PubMed.
Further Reading
Primary Papers
- Park L, Hochrainer K, Hattori Y, Ahn SJ, Anfray A, Wang G, Uekawa K, Seo J, Palfini V, Blanco I, Acosta D, Eliezer D, Zhou P, Anrather J, Iadecola C. Tau induces PSD95-neuronal NOS uncoupling and neurovascular dysfunction independent of neurodegeneration. Nat Neurosci. 2020 Sep;23(9):1079-1089. Epub 2020 Aug 10 PubMed.
Annotate
To make an annotation you must Login or Register.
Comments
The University of Minnesota
In this elegant paper, the authors demonstrated for the first time that the P301L/S mutation in tau attenuated the activity-dependent increase in blood flow to the brain. It is widely believed that cerebrovascular dysfunction in Alzheimer’s disease (AD) probably arises from cerebral amyloid angiopathy, in which amyloid-β deposits in the walls of small to medium blood vessels trigger the pathological blockade of these vessels. The present study instead provides a very novel and highly significant mechanistic link between tau abnormalities and cerebrovascular impairments, challenging the prevalent view.
One common mechanism for pathogenesis of neurodegenerative diseases involves the redistribution of the microtubule-associated protein tau from the axon into the somatodendritic compartment of neurons, followed by the mislocalization of tau into dendritic spines, resulting in postsynaptic functional deficits. The current study convincingly demonstrated that the disruption of the coupling between NMDA receptor and nitric oxide synthase by tau inhibited activity-triggered vasodilation. These results directly link postsynaptic activity to cerebrovascular dysfunction, providing a novel concept for how tau abnormalities can lead to some of the earliest pathophysiological changes during AD progression.…More
Nevertheless, the manuscript has not yet resolved the role of wild-type tau in angiopathy. Most AD patients do not have genetic mutations in tau. It will be interesting to know how changes in the expression level and postsynaptic modifications of tau contribute to the cerebrovascular dysfunction. Here, only transgenic-negative mice were used as the wild-type control. Additional experiments using transgenic mice expressing wild-type human tau proteins at different levels will likely present a clearer picture.
Mass General Institute for Neurodegenerative Disease
Park et al. use a combination of imaging methods including ASL-MRI and two-photon microscopy, as well as electrophysiology and biochemistry to examine neurovascular coupling in two lines of human-tau-overexpressing mice. Critically, they show that blood vessels in young tau mice fail to dilate properly and blood flow does not increase in response to a whisker-stimulation task, despite similar extents of neuronal activation when compared with wild-type controls. We had previously reported that, surprisingly, aged tau-overexpressing mice had abnormal blood vessels, including reduced overall capillary diameter, occlusion by leukocytes, and increasing tortuosity, which suggests impaired blood flow could contribute to ongoing processes of neurodegeneration (Bennett et al., 2018). By visualizing related changes in much younger mice, these data indicate that such blood flow abnormalities precede neuronal loss and thus may play a causal role.…More
This work is exciting, helping define a possible route of tau-mediated neurodegeneration and adding to a growing stack of evidence that tau and vasculature are intertwined. Disentangling this link could lead to new ideas in our understanding and treatment of Alzheimer’s disease: Are there specific pathologic soluble tau species that are responsible for disrupting neurovascular coupling and can they be targeted to improve disease outcome? Are there additional routes by which tau affects vasculature at different stages of the disease course? In following, are there points at which targeting tau may not provide the same benefits as seen when the authors suppressed tau expression early on in these mice? Importantly, is tau similarly affecting blood flow in human disease?
Altered BOLD-fMRI signal in Alzheimer’s hints that this could be the case and in an excellent talk at the AD/PD meeting in April this year, Michael Ewers presented tau PET data indicating regions with tau accumulation are associated with reduced cerebral blood flow. I look forward to seeing this area of science continue to grow and eagerly anticipate work from neuroimaging groups using combined PET/MRI techniques to assess similar relationships between tau and vasculature in human disease.
References:
Bennett RE, Robbins AB, Hu M, Cao X, Betensky RA, Clark T, Das S, Hyman BT. Tau induces blood vessel abnormalities and angiogenesis-related gene expression in P301L transgenic mice and human Alzheimer's disease. Proc Natl Acad Sci U S A. 2018 Feb 6;115(6):E1289-E1298. Epub 2018 Jan 22 PubMed.
Make a Comment
To make a comment you must login or register.