Scientists Suggest Curbing Sugar Modification to Tame BACE
Quick Links
One strategy scientists are trying to slow the progression of Alzheimer’s is inhibiting the BACE1 enzyme that cleaves the amyloid precursor protein (APP). However, since BACE1 cuts a range of other substrates, side effects are a concern. One group now reports that changing the sugar coating on the enzyme’s surface could tweak its function just enough to reduce APP cleavage, while perhaps sparing other substrates. Researchers led by Naoyuki Taniguchi, RIKEN-Max Planck Joint Research Center, Wako, Japan, report in the January 15 EMBO Molecular Medicine that knocking out a sugar transferase responsible for the modification reduced Aβ production and pathology in mice. The authors propose that inhibiting this enzyme could treat AD, and may pose fewer risks than blocking BACE1.
“This paper gives us a window onto a new avenue that we have not looked at carefully before,” said Gopal Thinakaran, University of Chicago. “Sugar modification of an enzyme could have a profound influence on the outcomes for Alzheimer’s pathogenesis.”

A Snug Fit.
The glycosyltransferase GnT-III, coded by the Mgat3 gene, inserts a monosaccharide GlcNAc at the fork in a sugar chain, bisecting it. [Image courtesy of Kizuka et al., 2015]
Glycosylation is a common post-translational modification of proteins. Added sugars enable everything from proper protein folding to ligand-receptor interactions. The sugars themselves can be further modified. For instance, the enzyme glycosyltransferase GnT-III (GnT-III) adds the monosaccharide N-acetylglucosamine (GlcNAc) in between the branches of a sugar called an N-glycan. This monosaccharide becomes a bisecting GlcNAc, being inserted at the junction of a bifurcating sugar chain (see image at left). GnT-III appears to be the only enzyme that can pull off this modification. Previous studies have suggested that bisecting GlcNAcs suppress cancer (see Song et al., 2010). However, their role in the brain remains unclear. Taniguchi’s group previously found higher amounts of GnT-III in the brains of people with AD than normal controls (see Akasaka-Manya et al., 2010). Since APP and its secretases carry sugars, the scientists wondered whether GnT-III adds bisecting GlcNAc to any of them, and how that might influence Aβ production.
First author Yasuhiko Kizuka and colleagues tested whether sugars on γ-secretase, β-secretase, or APP were decorated with bisecting GlcNAcs. They immunoprecipitated these proteins from APP23 mouse brains and blotted them with E4-PHA, a plant lectin that recognizes the bisecting GlcNAc structure. They found that neither γ-secretase nor APP reacted much, but BACE1 did. In APP23 mice lacking GnT-III, E4-PHA failed to recognize bisecting GlcNAcs on BACE1, suggesting that the enzyme modifies the secretase.
This one small sugar modification had a big impact on AD pathology. APP23 mice with no GnT-III had half the APP BACE1 cleavage products, lower levels of soluble and insoluble Aβ40 and Aβ42, and fewer than half the plaques in their brains, compared with animals with the enzyme. They also navigated a Y maze as well as controls. Together, the results imply that AD pathology worsens when GnT-III adds a bisecting GlcNAc onto BACE1.
Could the same could be true in AD patients? Kizuka reported elevated E4-PHA reactivity with BACE1 in the temporal lobe samples taken from postmortem brains of people who had died with early stage or later-stage AD.
How does the added sugar change BACE1? In vitro, bisecting GlcNAc had no effect on how well BACE1 cleaved its substrates. However, it did seem to relocate some of the enzyme from early endosomes to lysosomes. In brains from 3- and 12-month-old APP23 mice with functional GnT-III, BACE1 co-localized more with APP in early endosomes. In animals that lacked Gnt-III, about 5 percent less BACE1 hung around in endosomes, instead turning up in lysosomes. This hints that bisecting GlcNAc slows trafficking of BACE1 to lysosomes, making it more likely to cleave Aβ in endosomes, wrote the authors. Removing GnT-III cleared the enzyme a bit faster from endosomes (see image below).
The authors are unsure how this happens, but propose that an as-yet-unidentified protein recognizes the bisecting GlcNAc structure and directs the enzyme along the lysosomal pathway.
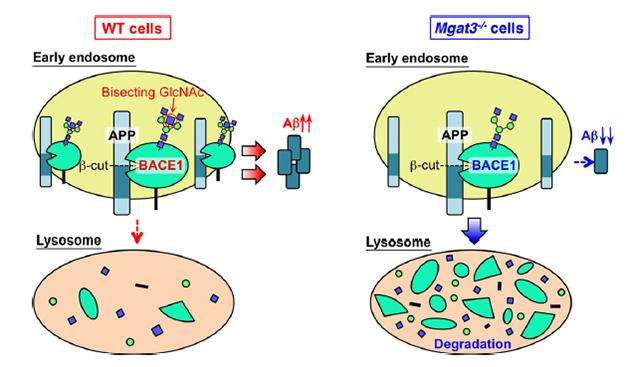
Sorting BACE1: A bisecting GlcNAc (red circle, left) keeps BACE1 in the early endosome, where it cleaves APP. Loss of the bisecting GlcNAc sends BACE1 to lysosomes, reducing Aβ generation (right). [Image courtesy of Kizuka et al., 2015.]
Interestingly, knocking out GnT-III did not affect BACE1 processing of the two other substrates, full-length CHL1 and contactin-2, whereas BACE inhibitors did block processing of both. While these are only two of the many known BACE1 substrates, the authors think that inhibiting GnT-III may specifically limit APP processing by BACE1. GnT-III knockouts have few significant deficiencies, therefore GnT-III could present a better drug target than BACE1, the authors contend (see Orr et al., 2013). No GnT-III inhibitor exists yet, but Taniguchi said that he and collaborators are preparing to screen 200,000 compounds.
“This is the first paper to describe an N-glycosylation of BACE1 that modifies its function,” said Cheng-Xin Gong, New York State Institute for Basic Research, Staten Island. He pointed out that GnT-III adds bisecting GlcNAcs to a number of other proteins, hence cautions that inhibiting it might have unexpected consequences, as well (see Zhao et al., 2008; Isaji et al., 2010).
Gong expressed doubt that altered trafficking accounts for the entire mechanism, given the small differences in lysosomal BACE between APP23 mice and their Mgat3-knockout counterparts. GnT-III could add the bisecting GlcNAc to the substrate-binding site for APP, he suggested, raising BACE1 affinity for the precursor protein. Thinakaran agreed that the differences between endosome and lysosome co-localization were small, and is likewise unconvinced that a shift in trafficking accounts for the mechanism. However, he agrees that something links the bisecting GlcNAc to APP processing, possibly through effects on BACE1.
In terms of therapeutics, Thinakaran was pleased that GnT-III inhibition did not affect other BACE1 substrates, but wondered if blocking GnT-III during adulthood might cause problems that do not occur in mice that lack GnT-III from the get-go.—Gwyneth Dickey Zakaib
References
Research Models Citations
Paper Citations
- Song Y, Aglipay JA, Bernstein JD, Goswami S, Stanley P. The bisecting GlcNAc on N-glycans inhibits growth factor signaling and retards mammary tumor progression. Cancer Res. 2010 Apr 15;70(8):3361-71. PubMed.
- Akasaka-Manya K, Manya H, Sakurai Y, Wojczyk BS, Kozutsumi Y, Saito Y, Taniguchi N, Murayama S, Spitalnik SL, Endo T. Protective effect of N-glycan bisecting GlcNAc residues on beta-amyloid production in Alzheimer's disease. Glycobiology. 2010 Jan;20(1):99-106. PubMed.
- Orr SL, Le D, Long JM, Sobieszczuk P, Ma B, Tian H, Fang X, Paulson JC, Marth JD, Varki N. A phenotype survey of 36 mutant mouse strains with gene-targeted defects in glycosyltransferases or glycan-binding proteins. Glycobiology. 2013 Mar;23(3):363-80. Epub 2012 Oct 31 PubMed.
- Zhao Y, Sato Y, Isaji T, Fukuda T, Matsumoto A, Miyoshi E, Gu J, Taniguchi N. Branched N-glycans regulate the biological functions of integrins and cadherins. FEBS J. 2008 May;275(9):1939-48. Epub 2008 Apr 1 PubMed.
- Isaji T, Kariya Y, Xu Q, Fukuda T, Taniguchi N, Gu J. Functional roles of the bisecting GlcNAc in integrin-mediated cell adhesion. Methods Enzymol. 2010;480:445-59. PubMed.
Further Reading
Papers
- Agostinho P, Pliássova A, Oliveira CR, Cunha RA. Localization and Trafficking of Amyloid-β Protein Precursor and Secretases: Impact on Alzheimer's Disease. J Alzheimers Dis. 2015;45(2):329-47. PubMed.
- Taniguchi N, Korekane H. Branched N-glycans and their implications for cell adhesion, signaling and clinical applications for cancer biomarkers and in therapeutics. BMB Rep. 2011 Dec;44(12):772-81. PubMed.
Primary Papers
- Kizuka Y, Kitazume S, Fujinawa R, Saito T, Iwata N, Saido TC, Nakano M, Yamaguchi Y, Hashimoto Y, Staufenbiel M, Hatsuta H, Murayama S, Manya H, Endo T, Taniguchi N. An aberrant sugar modification of BACE1 blocks its lysosomal targeting in Alzheimer's disease. EMBO Mol Med. 2015 Jan 15;7(2):175-89. PubMed.
Annotate
To make an annotation you must Login or Register.
Comments
Johns Hopkins University
UK Dementia Research Institute@UCL and VIB@KuLeuven
BACE1 stands as a very attractive therapeutic target for AD. However, with the identification of physiologically relevant BACE1 substrates, one should be aware that a complete block of BACE1 activity may lead to unintended side effects similar to what was seen with the γ-secretase inhibition studies, e.g., Semagacestat. Therefore, the discovery of alternative strategies to target these secretases are of high value.
The identification of this novel sugar modification of BACE1 that seems particularly increased in Alzheimer’s disease brain highlights once more the complexity of the processes going on during AD progression, and again calls for caution when rushing toward a new, poorly understood therapeutic approach.
The authors show that GnT-III, the enzyme responsible for the bisecting GlcNAc modification, is upregulated in the brains of AD patients, and suggest that this modification promotes AD pathogenesis by delaying BACE1 degradation. Although the authors claim that the bisecting GlcNAc blocks the lysosomal trafficking of BACE1 in the mouse brain, the difference in co-localization with the late endosome/lysosome marker Lamp1 is very small between the GnT-III-positive and –negative genotypes (~5 percent increase in the latter), which makes this argument rather weak. Nevertheless, it is clear that the bisecting GlcNAc modification modulates BACE1 levels and APP processing. Interestingly, the authors show that the inhibition of GnT-III inhibits only the processing of APP by BACE1, not the processing of the other substrates.
Since the GnT-III knockout mice do not present any obvious phenotypes, the authors emphasize the possibility of a new AD therapy by targeting this glycosyltransferase. Although very little is known about GlcNAc modification function, GlcNAc is highly expressed in the normal brain and it has been shown to suppress cancer metastasis. This evidence suggests that GlcNAc might play an important physiological role, and therefore the absence of a phenotype does not imply absence of risk for mechanistic side effects when blocking the enzyme. We should not forget that BACE1 knockout mice were also initially considered to be normal and throughout the years many complex phenotypes have been revealed.
Nevertheless, the discovery of this aberrant sugar modification is highly interesting and might inspire some researchers to look for alternative therapeutic strategies.
View all comments by Bart De StrooperUniversity of Connecticut Health
This study described a novel observation that BACE1 stability can be regulated through bisecting N-acetylglucosamine. The authors showed that BACE1 normally undergoes bisecting N-acetylglucosamine by glycosyltransferase GnT-III (encoded by the MGAT3 gene). Interestingly, MGAT3 levels were higher in AD patients, even noticeably higher in early AD patients’ brains than in non-demented controls. Consequently, more BACE1 was modified by MGAT3 as evidenced by the slightly slower migration on the SDS gel. When BACE1 is modified through bisecting N-acetylglucosamine, its stability is enhanced due to less trafficking to late endosomes/lysosomes for degradation. Their mouse genetic studies showed that mice lacking the MGAT3 gene decreased BACE1 stability by directing more BACE1 to the lysosome, and had a reduction in Aβ generation. This observation is important because inhibition of MGAT3 activity likely reduces amyloid deposition indirectly.
One unsolved physiological question is, why does MGAT3 target BACE1, but not APP, or other BACE1 substrates? It is known that cellular trafficking of BACE1 and APP are regulated by different pathways, although both are type I transmembrane proteins. Trafficking of BACE1 is also controlled by RTN3, and mice deficient in RTN3 show increased stability of BACE11. RTN3 interacts only with BACE1, not APP (see Shi et al., 2014).
References:
Shi Q, Ge Y, Sharoar MG, He W, Xiang R, Zhang Z, Hu X, Yan R. Impact of RTN3 deficiency on expression of BACE1 and amyloid deposition. J Neurosci. 2014 Oct 15;34(42):13954-62. PubMed.
View all comments by Riqiang YanMake a Comment
To make a comment you must login or register.