Research Brief: Crystal Clear—Prion-stabilizing Antibodies
Quick Links
Now hear this. There’s new support for the idea that antibodies that stabilize normal prion proteins offer hope of fighting Creutzfeldt-Jakob and other prion-based diseases. In the 9 February PNAS online, researchers led by John Collinge at the Medical Research Council Prion Unit, London, and S. Samar Hasnain at the Daresbury Laboratory, Warrington, England, detail x-ray crystallographic analysis of a prion-antibody complex. They found that a monoclonal antibody with high therapeutic potential binds with high affinity to normal prion (PrPc) rather than a toxic form (PrPSc). The finding may help researchers develop better antibodies for passive immunotherapy and helps explain why certain mutations in the prion proteins increase susceptibility to prion-based disease.
Prion diseases, which include mad cow disease (or bovine spongiform encephalopathy, BSE) and scrapie in sheep, are caused by the Dr. Jekyll/Mr. Hyde-like prions. The normally innocuous proteins assume a toxic form following a rare, though potentially lethal shape shift. Toxic PrPSc proteins form amyloids and also have a penchant for transmogrifying normal prions, setting off a self-perpetuating, neurodegenerative spiral. The bulk of evidence indicates that this self-catalytic conversion is what makes prions infectious and that stabilizing the normal PrPc might be a key to controlling prion diseases. The new research supports this idea.
First author Svetlana Antonyuk and colleagues tested monoclonal antibodies previously made against α- and β-PrP, which have the same basic conformations as PrPc and PrPSc, respectively. They found that antibodies that had the highest affinity for PrP were by far the most potent at inhibiting prion propagation in ScN2a neuroblastoma cells. The most potent antibody, ICSM 18, was previously shown to prevent prion disease in mice (see White et al., 2003).
To determine what kind of structure these antibodies recognize, the researchers took the Fab fragments (containing the antigen binding site) of the most potent antibody (ICSM 18) and crystallized it in the presence of a truncated form of recombinant human PrP (PrP119-231) containing the antibody binding site. That epitope is in residues 143-156, which make up helix H1 of the protein. That α helix is thought to contribute to prion toxicity by undergoing a conformational change to β-sheet structure that helps drive conversion to PrPSc. The crystal structure shows a significant stabilization of the H1 helix. It also shows that amino acids 197-205, which form part of a disordered segment of human PrPc that may promote β-sheet formation, are in close proximity to the heavy chain of the antibody. “The physical proximity of the Fab H chain to a region of PrP that is a possible site for β-sheet formation suggests that the complex may provide structural inhibition by burying the ‘active’ residues at this interface,” write the authors (see image below).
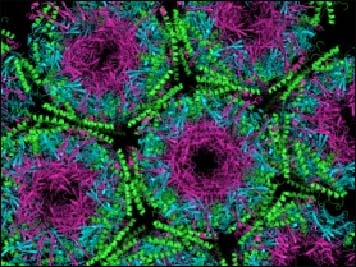
A Packed House
The crystal structure shows light (magenta) and heavy (cyan) chains of the monoclonal antibody packed together with normal PrP (green). Image credit: Samar Hasnain, University of Liverpool, U.K.
The 1-to-2 antibody-to-prion complex further shows an interaction between two β-sheet strands (amino acids 129-131 and 161-163) that have been found sidling up to each other in structures obtained in the absence of antibody (see Haire et al., 2004). The authors suggest that this is hardly coincidence and may be related to prion propagation. That idea is supported by the fact that polymorphisms in amino acid 129 have a dramatic effect on prion transmission and pathogenesis. Heterozygosity at this position is protective against prion disease in humans, while transmission of BSE from cows to humans appears to occur exclusively in people who are homozygous for methionine at that position. “Heterozygosity is protective owing to the requirement for sequence homogeneity in forming ordered, self-replicating particles,” suggest the authors.
In other prion news this week, researchers led by Stanley Prusiner at the University of California, San Francisco, report that different polyoxometalates (POMs), such as phosphotungstate anions, influence whether prions form into amyloid-like rods or two-dimensional crystals. It is not clear by what mechanism POMs work their magic, or if there may be POMs that can be used to create 3D crystals for ultrastructural analysis. The authors do not address whether POMs might offer any therapeutic value.—Tom Fagan
References
Paper Citations
- White AR, Enever P, Tayebi M, Mushens R, Linehan J, Brandner S, Anstee D, Collinge J, Hawke S. Monoclonal antibodies inhibit prion replication and delay the development of prion disease. Nature. 2003 Mar 6;422(6927):80-3. PubMed.
- Haire LF, Whyte SM, Vasisht N, Gill AC, Verma C, Dodson EJ, Dodson GG, Bayley PM. The crystal structure of the globular domain of sheep prion protein. J Mol Biol. 2004 Mar 5;336(5):1175-83. PubMed.
Further Reading
Primary Papers
- Antonyuk SV, Trevitt CR, Strange RW, Jackson GS, Sangar D, Batchelor M, Cooper S, Fraser C, Jones S, Georgiou T, Khalili-Shirazi A, Clarke AR, Hasnain SS, Collinge J. Crystal structure of human prion protein bound to a therapeutic antibody. Proc Natl Acad Sci U S A. 2009 Feb 24;106(8):2554-8. PubMed.
- Wille H, Shanmugam M, Murugesu M, Ollesch J, Stubbs G, Long JR, Safar JG, Prusiner SB. Surface charge of polyoxometalates modulates polymerization of the scrapie prion protein. Proc Natl Acad Sci U S A. 2009 Mar 10;106(10):3740-5. PubMed.
Annotate
To make an annotation you must Login or Register.
Comments
GSBio, LLC
Protein misfolding and/or aggregation is a common characteristic underlying many neurodegenerative diseases. One approach for treating these diseases is with therapeutics that target the disease protein in question, either by interfering with production or propagation of its disease specific isoform/conformation, or by promoting its clearance. Antibodies have found particular utility as therapeutic agents in this regard, most notably in AD and prion disease. At the same time, it is widely appreciated that not all antibodies to a particular target have equivalent activities. Recently, investigators have begun turning to x-ray crystallography to provide greater insight at the atomic level into features that distinguish antibodies with differing in-vitro or in-vivo activities, to guide approaches for improved therapeutics, and ultimately, to gain insights into structural features of the disease protein which contribute to its pathologic form. This study by Antonyuk et al. provides a very nice illustration of how structural analysis benefits these objectives.
The investigators provide evidence that antibody potency (defined as neutralizing activity in cell models of antibodies recognizing two in-vitro generated conformations of PrPc, termed αPrP and βPrP), correlates with their binding affinities for PrPc, particulary for cell surface exposed epitopes of PrPc. Antonyuk et al. investigated the structural basis underlying the bioactivity of one of their more in-vitro and in-vivo potent antibodies, ICSM 18, using x-ray crystallography of a truncated PrPc fragment (residues 119-231), in complex with a Fab fragment of the antibody. The x-ray structure revealed two key details: a) numerous contact points between the antibody and a helical segment, termed H1 (residues 143-156), of the PrPc fragment; and b) a series of 4 anti-parallel β-sheets that mediate interactions between neighboring PrPc molecules. The helical segment has been implicated in the β-sheet transformation that distinguishes PrPSc from PrPc, and tight binding of this domain in PrPc by ICSM 18, via the multiple contacts, is consistent with the bioactivity of ICSM 18, i.e., it stabilizes PrPc. Independently, the anti-parallel β-sheets have also been observed between neighboring PrP molecules in the crystal structure of sheep PrP, and the authors speculate that a critical residue at position 129 in this region may explain genetic susceptibility and prion strain selection in humans by contributing to PrPSc formation, and prion propagation. Hence, the study of Antonyuk et al. highlights a preferred epitope on PrPc for therapeutic mAbs, and suggests how the structural features elucidated provide a basis for prion disease. The implication of their finding regarding the epitope bound by ICSM 18 would benefit from comparative x-ray crystallography of PrPc complexed with an antibody that has equal binding affinity as ICSM 18, but lower potency for inhibiting prion propagation in cells of, e.g., ICSM antibodies 17, or 19.
Make a Comment
To make a comment you must login or register.