From Protector to Instigator, Autophagy Makes About-Face in ALS
Quick Links
Autophagy: good or bad? The answer may depend on when you're asking about. According to a September 11 paper in Proceedings of the National Academy of Sciences, motor neurons use this “self-eating” function to stave off synaptic malfunction early in the course of a mouse model of amyotrophic lateral sclerosis (ALS). But as the disease progresses, motor neuron autophagy somehow ignites harmful responses in other cells, including interneurons and glia. The result of this two-faced pathway? In ALS mice lacking autophagy in motor neurons, symptoms arose earlier and yet the animals lived longer. The study, led by Tom Maniatis at Columbia University in New York, suggests caution is in order for those developing autophagy-based therapies for ALS.
- In SOD1-G93A ALS mice, scientists deleted the autophagy gene Atg7 in motor neurons.
- These mice developed ALS at a younger age but lived longer than SOD1-G93A controls.
- Early on, autophagy in motor neurons delayed denervation of neuromuscular junctions, but later, it stressed interneurons and inflamed glia.
“This study is compelling,” commented David Rubinsztein of the University of Cambridge in England. “Factors determining onset may differ from those that affect progression. This may be important when designing therapies.”
Best known for its role in dispatching unwanted proteins and organelles, autophagy is the process by which a double membrane compartment engulfs cytoplasmic detritus and fuses with lysosomes to digest it (for review, see Guo et al., 2017). Autophagy also plays a part in numerous other neuronal functions, including synaptic remodeling and neurotransmission (Hernandez et al., 2012; Inoue et al., 2013).
Mutations in multiple autophagy genes are implicated in ALS. Furthermore, even motor neurons from patients without such mutations are chock-full of inclusions of p62, a receptor that delivers proteins to autophagosomes (Mizuno et al., 2006). These observations place autophagy at the heart of both familial and sporadic forms of ALS. However, what autophagy does in ALS is anything but clear. For example, despite its ability to clear pathogenic protein aggregates (a good thing), one study reported that the autophagy activator, rapamycin, shortened the lifespan of ALS model mice, and another found that genetically hobbling autophagy extended the animals’ lives (Zhang et al., 2011; Nassif et al., 2014). Because autophagy was altered in many cell types in these and other studies, it remained unclear how the pathway was influencing disease progression.

Flavors of Inclusion.
In wild-type animals, p62 spreads diffusely throughout motor neurons (left). The autophagy receptor forms round bodies (middle) and skein-like inclusions (right) in SOD1-G93A mice. [Courtesy of Rudnick et al., PNAS, 2017.]
First author Noam Rudnick and colleagues decided to take examine how the pathway’s influence changed throughout the course of ALS and in different cell types. They first assessed autophagy markers in the spinal cord of the SOD1-G93A model. In the asymptomatic phase of the disease, when the animals were 50 days old, the researchers found that p62 inclusions came in two distinct breeds: round bodies (RBs) in neuronal cell bodies, and skein-like inclusions (SKIs), which took up residence in the soma and dendrites (see image above). RBs predominated early on, but by 150 days, SKIs were more abundant. Fluorescence staining revealed that fast motor neurons—the cells most vulnerable to neurodegeneration in ALS—were the primary bearers of RBs, while SKIs appeared in slow motor neurons and later in interneurons as well.
A closer comparison of the inclusions in motor neurons revealed that while RBs were bona fide mature autophagosomes, SKIs lacked mature autophagosome markers and contained aggregates of mutant SOD1. The researchers hypothesized that this build-up of toxic proteins without recruitment of the appropriate digestion machinery might stress SKI-containing cells. They were right: SKI-bearing motor neurons had an abundance of phosphorylated cJun, a marker of cell stress, in their nuclei.
To better understand how different types of neurons use autophagy, the researchers conditionally knocked out Atg7, an enzyme required to make autophagosomes, only in motor neurons. In these autophagy-deficient but otherwise normal mice, p62 accumulated dramatically in motor neurons, which swelled in size but did not degenerate. Though the cells survived, a fraction of fast motor neurons lost connections with muscles at neuromuscular junctions (NMJs), and pumped out fewer synaptic vesicles than those in normal mice. The researchers concluded that fast motor neurons depend on autophagy to innervate muscles, but not for survival.
To investigate how loss of autophagy in motor neurons would affect disease, the researchers generated double transgenic mice that lacked Atg7 in motor neurons, and also expressed SOD1-G93A. Hind-limb tremor, the first motor symptom of disease, emerged 22 percent earlier in these double transgenic mice than in SOD1-G93A mice, though both single and double transgenic mice started losing weight around the same time. Surprisingly, knocking out autophagy in motor neurons extended the lifespan of SOD1-G93A mice by 22 percent. Both types of mice had similar numbers of motor neurons at the early and late stages. Interestingly, while motor neurons started disconnecting from NMJs much sooner in the double transgenics, they ended up with more intact NMJs than their autophagy-competent counterparts by 150 days. The findings suggested that autophagy benefits motor neurons at first, but later promotes harmful denervation that accelerates disease.
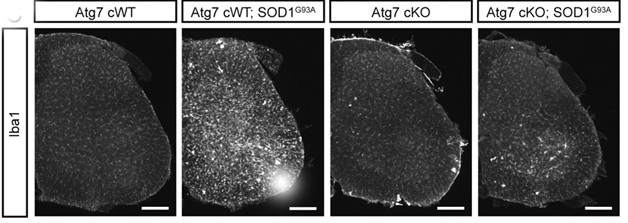
Autophagy, the Instigator. Activated microglia (Iba1, white) infiltrate the spinal cords of SOD1-G93A mice (second panel), but not when motor neurons cannot perform autophagy (fourth panel). [Courtesy of Rudnick et al., PNAS, 2017.]
How was autophagy accelerating disease progression? Previous studies have suggested that damaging neuroinflammatory responses take hold in the later stages of ALS, so the researchers compared activation of glial cells in the different mouse models. Indeed, SOD1-G93A mice had inflamed spinal cords at day 150, with numerous activated astrocytes and microglia. This was dramatically reduced in SOD1-G93A mice lacking Atg7 in motor neurons, suggesting that autophagy in motor neurons somehow triggers inflammatory responses in glia. Interneurons were affected by the loss of autophagy in motor neurons, as well. These in-betweeners lacked the p62-loaded SKIs and the nuclear cJun expression observed in animals with autophagy-competent motor neurons. Though delayed, both glial inflammation and interneuron stress eventually cropped up in the Atg7-deficient SOD-G93A animals.
How motor neuron autophagy incites harmful reactions in other cells remains a mystery. One possibility is that autophagy sends an activation signal to other cells, possibly when the disposal pathway becomes overwhelmed, the researchers proposed. Robert Baloh of Cedars-Sinai Medical Center in Los Angeles agreed, yet called the idea counterintuitive. “You would think that if motor neurons lacked autophagy, and were thus filled with inclusions that could not be cleared, that would cause more stress in other cells,” he said. “But instead, maybe there is a specific signal from autophagy that needs to be released to get glial reaction.”
Baloh said the findings should caution researchers who see autophagy activation as a potential therapeutic strategy. “Much like the story with neuroinflammation, a lot depends on when you activate it,” he said.
Johnathan Cooper-Knock of the University of Sheffield in England wondered how the timeline in mice corresponded to human disease. “It is interesting to speculate at what stage of human ALS a switch may occur from predominant fast motor neuron pathology to non-cell autonomous toxicity driven indirectly by dying fast motor neurons which have performed autophagy on pathological aggregates,” he wrote in an email to Alzforum. “This may be as early as onset of clinical symptoms—certainly neuroinflammation and gliosis is universally reported in human ALS.” He added that the results will need to be replicated in other animal models of ALS beyond SOD1-G93A.
The results dovetail with work led by Claudio Hetz of the University of Chile in Santiago, who reported that activating autophagy in an ALS model mice triggered stress responses. “These results confirm our previous observations indicating that in the context of ALS, the autophagy and ER stress pathways are interconnected in a dynamic way, and that reducing autophagy levels may be beneficial,” he wrote in an email to Alzforum.—Jessica Shugart
References
Research Models Citations
Paper Citations
- Guo F, Liu X, Cai H, Le W. Autophagy in neurodegenerative diseases: pathogenesis and therapy. Brain Pathol. 2018 Jan;28(1):3-13. Epub 2017 Aug 6 PubMed.
- Hernandez D, Torres CA, Setlik W, Cebrián C, Mosharov EV, Tang G, Cheng HC, Kholodilov N, Yarygina O, Burke RE, Gershon M, Sulzer D. Regulation of presynaptic neurotransmission by macroautophagy. Neuron. 2012 Apr 26;74(2):277-84. PubMed.
- Inoue K, Rispoli J, Yang L, MacLeod D, Beal MF, Klann E, Abeliovich A. Coordinate Regulation of Mature Dopaminergic Axon Morphology by Macroautophagy and the PTEN Signaling Pathway. PLoS Genet. 2013 Oct;9(10):e1003845. PubMed.
- Mizuno Y, Amari M, Takatama M, Aizawa H, Mihara B, Okamoto K. Immunoreactivities of p62, an ubiqutin-binding protein, in the spinal anterior horn cells of patients with amyotrophic lateral sclerosis. J Neurol Sci. 2006 Nov 1;249(1):13-8. PubMed.
- Zhang X, Li L, Chen S, Yang D, Wang Y, Wang Z, Le W. Rapamycin treatment augments motor neuron degeneration in SOD1(G93A) mouse model of amyotrophic lateral sclerosis. Autophagy. 2011 Apr;7(4):412-25. PubMed.
- Nassif M, Valenzuela V, Rojas-Rivera D, Vidal R, Matus S, Castillo K, Fuentealba Y, Kroemer G, Levine B, Hetz C. Pathogenic role of BECN1/Beclin 1 in the development of amyotrophic lateral sclerosis. Autophagy. 2014 Jul;10(7):1256-71. Epub 2014 May 12 PubMed.
Further Reading
Primary Papers
- Rudnick ND, Griffey CJ, Guarnieri P, Gerbino V, Wang X, Piersaint JA, Tapia JC, Rich MM, Maniatis T. Distinct roles for motor neuron autophagy early and late in the SOD1G93A mouse model of ALS. Proc Natl Acad Sci U S A. 2017 Sep 26;114(39):E8294-E8303. Epub 2017 Sep 13 PubMed.
Annotate
To make an annotation you must Login or Register.
Comments
This study has added more complexity to the field, and suggests a new concept that should be considered for future therapeutic strategies to target autophagy in ALS. Despite initial expectations that blocking autophagy should exacerbate the severity of the diseases, the authors provide elegant data indicating that the opposite is true.
These results may reflect the fact that autophagy is deregulated in ALS, associated with an over-activation that is detrimental to motor neuron function. But also, the authors show that blocking autophagy restores some of the proteostasis defects observed in the disease, suggesting a complex crosstalk between different pathways that maintain the health of the proteome.…More
These results also confirm our previous observations indicating that, in the context of ALS, the autophagy and ER stress pathways are interconnected in a dynamic way, and that reducing autophagy levels may be beneficial. With this study, it is now becoming clear that the field needs to perform systematic studies to define the significance of autophagy to specific diseases one by one, since in certain contexts stimulating the pathway may be beneficial whereas in others it may be detrimental.
University of Sheffield
Rudnick and colleagues have performed a very elegant study of autophagy in the mtSOD1 model of ALS, with some surprising results. Selective vulnerability of motor neurons has long been noted in patients and in animal models, but not understood. Independent of ALS, the authors examined Atg7 cKO mice, which have disrupted autophagy. They demonstrated that dependence on autophagy to clear ubiquitinated aggregates appears to be specific to the “fast” motor neurons, which are most vulnerable to ALS-neurodegeneration. Interestingly, these mice exhibited denervation of fast-twitch muscle fibres without overt cell death.
This mimics the earliest phase of human ALS, and therefore it was perhaps unsurprising that crossing these mice with mtSOD1 mice, which develop ALS, accelerated the progression of early denervation related to ALS.…More
Where this study really breaks new ground is the observation that, despite accelerated denervation in early disease, the double transgenic mice survived longer and displayed reduced levels of neuronal loss and neuroinflammation in the late phase of disease. The authors suggest that this is because the late phase of disease may be driven by pathological material taken up in autophagosomes by fast motor neurons.
Much speculation has occurred around the idea that misfolded protein may spread disease through a “prion-like” mechanism, and these data would be consistent with that idea. The conclusion of this study is that therapeutic strategies aimed at increasing autophagy of pathological aggregates may need to be careful not to accelerate toxicity by other pathways.
Some questions remain: this study has focused on the well-characterised mtSOD1 model of ALS and will require independent validation to demonstrate that it is applicable more broadly to non-mtSOD1 ALS. It is also interesting to speculate at what stage of human ALS a switch may occur from predominant fast motor neuron pathology to non-cell autonomous toxicity driven indirectly by dying fast motor neurons which have performed autophagy on pathological aggregates. This may be as early as onset of clinical symptoms—certainly neuroinflammation and gliosis is universally reported in human ALS.
Make a Comment
To make a comment you must login or register.