Nixing Neuron Receptor Improves Recovery from Stroke, Trauma
Quick Links
Recovery from stroke can be slow-going. Neurons struggle to sprout new axons, make new dendritic spines, and form new synapses. Is there any way, besides standard physical therapy, to help the process along? A February 21 paper in Cell suggests a possible molecular drug target, the C-C chemokine receptor 5 (CCR5). Scientists led by S. Thomas Carmichael, University of California, Los Angeles, report that the receptor pops up on neurons after stroke or traumatic brain injury in mice, and that either silencing the gene or blocking the receptor with an FDA-approved drug enhanced recovery. Interestingly, people with a loss-of-function mutation in CCR5 also recover faster from stroke. The results could lead the way to a stroke treatment for people. “There currently is no drug that regenerates neurons after stroke,” Carmichael told Alzforum. Because the receptor also suppresses memory in normal mice, it could be a target for dementia as well, he said.
- Mouse neurons express the CCR5 receptor post-stroke.
- People with a mutation in CCR5 recover better from stroke.
- Knocking down or blocking the receptor improves motor function and learning and memory in mice with brain trauma.
Just over two years ago, co-author Alcino Silva, also at UCLA, found that mouse CCR5 knockouts demonstrate enhanced memory in several tasks (Zhou et al., 2016). As stroke recovery often recruits molecular and cellular mechanisms that are utilized during learning and memory, Carmichael wondered if knocking down CCR5 could speed the process.
To find out, first author Mary Joy and colleagues examined CCR5 expression on neurons. The receptor was known to be expressed on microglia, but its appearance on other cell types hadn’t been described. In normal mice, Joy found abundant CCR5 on microglia only, not on neurons. However, 12 hours after inducing a blood clot in a small area of the cortex, CCR5 levels on neurons near the infarct jumped, even surpassing amounts seen on microglia. Without intervention, that elevation lasted at least a month.
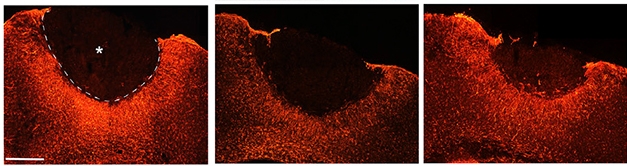
Calming astrocytes. Seven days after stroke, reactive astrocytes (red) surround the stroke area (asterisk with dotted border) in control mice (left). Reduced activity surrounds the infarct in animals treated with maraviroc (middle) or a gene-silencing regimen (right). [Image courtesy of Joy et al., Cell.]
To silence CCR5 post-stroke, the authors injected cortices of the mice with a neuron-targeting AAV virus coding a small hairpin RNA (shCCR5). The injection was timed so that downregulation of CCR5 didn’t happen until a few days after the stroke, allowing the initial upregulation of CCR5. Joy tracked the animals for nine weeks after the stroke.
The AAV-treated animals recovered more fully than did untreated littermates. Within a week, treated mice fell through a mesh grid half as often as controls. By this time, too, they supported their weight on both forelimbs, as opposed to relying mostly on the unaffected side. These benefits lasted the nine weeks of the experiment.
As it happens, CCR5 is important for HIV infection. One high-risk individual who never succumbed to the virus turned out to have a loss-of-function mutation in this gene. Subsequently, the FDA approved maraviroc, which blocks the receptor, for HIV patients. Would maraviroc have similar effects to CCR5 knockdown in the stroke models? The researchers gave mice the drug daily for nine weeks beginning 24 hours after a stroke. Within three weeks, the animals walked and supported their weight similarly to the AAV-shCCR5-treated mice, and the benefits also lasted at least nine weeks. A three-week stint on the drug worked just as well.
Blocking the receptor also helped mice recover after a traumatic brain injury. When AAV-shCCR5 was injected into their hippocampi two weeks before, or maraviroc was given to the mice just after a small weight was dropped on the skull, the animals regained learning and memory faster than untreated TBI controls. They explored a novel object for just as long as uninjured controls, and found a target hole in a circular Barnes maze just as quickly.
To explore the mechanism by which CCR5 knockdown improved recovery, the researchers analyzed neuronal protein levels a week after a stroke. Neurons from mice that had been treated with the AAV-shCCR5 increased expression of both dual leucine zipper kinase (DLK) and CREB. CREB previously had been found to be important in axonal sprouting and stroke recovery (Caracciolo et al., 2018; Moore and Goldberg, 2011). DLK has also been tied to axonal sprouting and axonal regeneration (Watkins et al., 2013). Joy and colleagues also saw that more existing dendritic spines survived post-stroke in animals without the receptor or given maraviroc, and more new spines were created.

Schematic. After a stroke (black spot on brain), CCR5 expression on neurons ramps up. Blocking it improves movement and learning and memory in mice, while people with a CCR5 deletion recover better as well (top). In mice, CCR5 blockade preserves dendritic spines, increases DLK and CREB signaling, and leads to axonal sprouting (bottom). [Courtesy of Joy et al., Cell.]
CCR5 knockdown also appeared to have effects on the immune system. A week after stroke, knockdown quieted astrocytic reactivity in areas surrounding the infarct compared with untreated animals (see image above). Knockdown also caused the brain to recruit fewer peripheral monocytes and macrophages to the stroke site.
To investigate whether any of this is relevant to people, Carmichael and colleagues turned to the Tel Aviv Brain Acute Stroke Cohort (TABASCO), which has enrolled and studied outcomes of 446 stroke patients. Of those, 68 people are carriers of the CCR5-Δ32 deletion, which causes a loss of CCR5 function. A year after stroke, carriers had recovered motor, language, and sensory capabilities better than noncarriers. They also tested higher on measures of memory, verbal function, attention, and total cognition. This is the first report of a human genetic variant that enhances healing from stroke, wrote the authors.
Taken together, these results suggest that the uptick in neuronal CCR5 interferes with stroke recovery, the authors wrote. Carmichael speculated that the receptor plays a protective role at first, by dampening excitability in the brain to limit damage. However, prolonged upregulation of this receptor appears to be deleterious, he said.
“It’s interesting how everything came together here,” Carmichael told Alzforum. “We have established a mechanism of action, we have an FDA-approved drug that works in mice and is safe in humans, and the TABASCO study suggests the receptor has a role in human recovery.” Based on all this, Carmichael and colleagues have initiated a Phase 2 trial that will test maraviroc in stroke patients. About 30 people will receive the drug or placebo about four to six weeks after stroke to see if the drug helps rehabilitation.
Are there implications for Alzheimer’s disease? Silva is excited about the possibility. “Because aging increases both memory deficits and the probability of strokes—including subclinical strokes that may exacerbate Alzheimer’s—blocking the receptor may help age-related cognitive decline and memory deficits associated with animal models of Alzheimer’s,” he wrote to Alzforum. He plans to examine aging mice and mouse models of AD to see if CCR5 is upregulated on neurons. If so, he’ll test whether knockdown or maraviroc treatment prevents or treats deficits in learning and memory. Small studies conducted in Iran and Europe suggest CCR5-Δ32 carriers are not protected against AD (Khorram Khorshid et al., 2012; Galimberti et al., 2004).
On that note, the world learned late last year that the CCR5 gene was CRISPR-edited and knocked out in a pair of twin girls born in China. As widely reported in The New York Times and other outlets, Jiankui He, Southern University of Science and Technology, Shenzhen, reportedly wanted to make the girls resistant to the HIV virus, which their father had. How that editing might affect human brain development and/or learning and memory is unknown.—Gwyneth Dickey Zakaib
References
Paper Citations
- Zhou M, Greenhill S, Huang S, Silva TK, Sano Y, Wu S, Cai Y, Nagaoka Y, Sehgal M, Cai DJ, Lee YS, Fox K, Silva AJ. CCR5 is a suppressor for cortical plasticity and hippocampal learning and memory. Elife. 2016 Dec 20;5 PubMed.
- Caracciolo L, Marosi M, Mazzitelli J, Latifi S, Sano Y, Galvan L, Kawaguchi R, Holley S, Levine MS, Coppola G, Portera-Cailliau C, Silva AJ, Carmichael ST. CREB controls cortical circuit plasticity and functional recovery after stroke. Nat Commun. 2018 Jun 8;9(1):2250. PubMed.
- Moore DL, Goldberg JL. Multiple transcription factor families regulate axon growth and regeneration. Dev Neurobiol. 2011 Dec;71(12):1186-211. PubMed.
- Watkins TA, Wang B, Huntwork-Rodriguez S, Yang J, Jiang Z, Eastham-Anderson J, Modrusan Z, Kaminker JS, Tessier-Lavigne M, Lewcock JW. DLK initiates a transcriptional program that couples apoptotic and regenerative responses to axonal injury. Proc Natl Acad Sci U S A. 2013 Mar 5;110(10):4039-44. Epub 2013 Feb 19 PubMed.
- Khorram Khorshid HR, Manoochehri M, Nasehi L, Ohadi M, Rahgozar M, Kamali R. Ccr2-64i and Ccr5 Δ32 Polymorphisms in Patients with Late-Onset Alzheimer's disease; A Study from Iran (Ccr2-64i And Ccr5 Δ32 Polymorphisms in Alzheimer's disease). Iran J Basic Med Sci. 2012 Jul;15(4):937-44. PubMed.
- Galimberti D, Fenoglio C, Lovati C, Gatti A, Guidi I, Venturelli E, Cutter GR, Mariani C, Forloni G, Pettenati C, Baron P, Conti G, Bresolin N, Scarpini E. CCR2-64I polymorphism and CCR5Delta32 deletion in patients with Alzheimer's disease. J Neurol Sci. 2004 Oct 15;225(1-2):79-83. PubMed.
External Citations
Further Reading
Papers
- Overman JJ, Clarkson AN, Wanner IB, Overman WT, Eckstein I, Maguire JL, Dinov ID, Toga AW, Carmichael ST. A role for ephrin-A5 in axonal sprouting, recovery, and activity-dependent plasticity after stroke. Proc Natl Acad Sci U S A. 2012 Aug 14;109(33):E2230-9. Epub 2012 Jul 25 PubMed.
- Di Lazzaro V, Profice P, Pilato F, Capone F, Ranieri F, Pasqualetti P, Colosimo C, Pravatà E, Cianfoni A, Dileone M. Motor cortex plasticity predicts recovery in acute stroke. Cereb Cortex. 2010 Jul;20(7):1523-8. Epub 2009 Oct 5 PubMed.
News
- Tregs Suppress Astrogliosis and Improve Recovery After Stroke
- Tau Toxicity Blamed for Widespread Cell Death After Stroke
- Using Clinical Dx Only, Study Calls Stroke Clear Risk Factor for LOAD
- Do Mild Traumatic Brain Injuries Double the Risk of Dementia?
- Genetic Resilience—Can We Find Treatments for the Sick by Studying the Healthy?
Primary Papers
- Joy MT, Ben Assayag E, Shabashov-Stone D, Liraz-Zaltsman S, Mazzitelli J, Arenas M, Abduljawad N, Kliper E, Korczyn AD, Thareja NS, Kesner EL, Zhou M, Huang S, Silva TK, Katz N, Bornstein NM, Silva AJ, Shohami E, Carmichael ST. CCR5 Is a Therapeutic Target for Recovery after Stroke and Traumatic Brain Injury. Cell. 2019 Feb 21;176(5):1143-1157.e13. PubMed.
Annotate
To make an annotation you must Login or Register.
Comments
Icahn School of Medicine at Mount Sinai
The report on CCR5 provides prima facie evidence of the long-held notion that much of the damage to neurons in brain disease occurs not as a result of the primary event, but due to activation of signaling by cytokines and other transmitters. This dovetails well with the recent network computational analysis by Emilie Castranio and colleagues at the University of Pittsburgh, who studied brain post-TBI and discovered that the signaling network centered around the TYROBP/DAP12 hub (driver) was selectively perturbed (Castranio et al., 2017).
TYROBP/DAP12 is the transmembrane adaptor responsible for signaling across TREM2 and CR3 and passing signals along to Syk protein kinase. C1q seems to be essential in this process, as shown by Mickael Audrain, Jean-Vianney Haure-Mirande and their colleagues at the Icahn School of Medicine at Mount Sinai in their back-to-back papers in press in the early online electronic publication section of Molecular Psychiatry (Audrain et al. 2018; Haure-Mirande et al., 2018).
What is also impressive is the ability of neurons to express molecules previously considered to be strictly the purview of inflammatory and immune cells. This creates enormous complexity in attempting to predict which immune inflammatory molecules are most important at which stages and in which cells.
This new CCR5 paper is groundbreaking in that the benefit is already established, and now Joy et al. and others can focus on working backward to discover the molecular underpinnings.
References:
Castranio EL, Mounier A, Wolfe CM, Nam KN, Fitz NF, Letronne F, Schug J, Koldamova R, Lefterov I. Gene co-expression networks identify Trem2 and Tyrobp as major hubs in human APOE expressing mice following traumatic brain injury. Neurobiol Dis. 2017 Sep;105:1-14. Epub 2017 May 11 PubMed.
Audrain M, Haure-Mirande JV, Wang M, Kim SH, Fanutza T, Chakrabarty P, Fraser P, St George-Hyslop PH, Golde TE, Blitzer RD, Schadt EE, Zhang B, Ehrlich ME, Gandy S. Integrative approach to sporadic Alzheimer's disease: deficiency of TYROBP in a tauopathy mouse model reduces C1q and normalizes clinical phenotype while increasing spread and state of phosphorylation of tau. Mol Psychiatry. 2019 Sep;24(9):1383-1397. Epub 2018 Oct 3 PubMed.
Haure-Mirande JV, Wang M, Audrain M, Fanutza T, Kim SH, Heja S, Readhead B, Dudley JT, Blitzer RD, Schadt EE, Zhang B, Gandy S, Ehrlich ME. Correction: Integrative approach to sporadic Alzheimer's disease: deficiency of TYROBP in cerebral Aβ amyloidosis mouse normalizes clinical phenotype and complement subnetwork molecular pathology without reducing Aβ burden. Mol Psychiatry. 2019 Mar;24(3):472. PubMed.
Make a Comment
To make a comment you must login or register.