Nicastrin Bounces Bulky Proteins from γ-Secretase
Quick Links
Nicastrin has been proposed to help the γ-secretase complex select which transmembrane proteins to cut, but the exact mechanism remains controversial. New data from scientists led by Michael Wolfe at Brigham and Women's Hospital, Boston, suggests that nicastrin blocks substrates with large ectodomains from accessing the enzyme’s active site, allowing only shorter substrates to be cleaved. The model questions previous theories that nicastrin actively binds and recruits substrates for γ-secretase, and may explain why α- or β-secretase must first shed most of the substrate ectodomain before γ cleavage can occur, helping scientists better understand Aβ regulation. The researchers published their data in the December 22 Proceedings of the National Academy of Sciences.

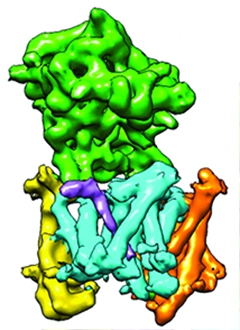
Keeping it Simple.
Nicastrin (green) perches above the plasma membrane (gray lines), blocking proteins with large ectodomains (orange) from the core of the g-secretase complex (multicolored). [Image courtesy of David Bolduc, PNAS.]
Nicastrin is a type-I transmembrane protein that forms a complex with three other proteins to form the γ-secretase complex (see image at left). Presenilin, the catalytic subunit, only cleaves substrates that have had their ectodomains mostly sheared off by α- or β-secretase. The substrates of the protease include Notch, crucial for cell fate determination, and APP, important for Alzheimer’s disease (AD).
Previous reports suggested nicastrin actively recruits substrates. A decade ago, Gang Yu and colleagues at the University of Texas Southwestern Medical Center, Dallas, proposed that glutamate at position 333 of nicastrin binds the substrate’s ectodomain stub left behind after α- or β-secretase cleavage, and helps pull the substrate close to the active site (Shah et al., 2005). However, recent high-resolution structures place E333 deep inside nicastrin and distant from the active site, causing scientists to question how it could bind substrates (see Aug 2015 news; May 2015 news). What’s more, first author David Bolduc pointed out that γ-secretase has more than 90 substrates with diverse ectodomain sequences, and the likelihood that they contain a common recognition sequence is small. Could nicastrin select for substrates a different way?
To find out, Bolduc and colleagues used E. coli to produce a truncated version of Notch that resembles the fragment produced after cleavage by α-secretase. In vitro, when the researchers varied the amino acid at the N-terminal tip of the truncated ectodomain, or lopped it off entirely, γ-secretase cut Notch with the same efficiency. This refuted the idea that the ectodomain harbored a specific site that flagged it as a substrate for γ-secretase. However, if the scientists lengthened the substrate’s ectodomain, either by extending its amino acids or tacking on a whole other protein region, γ-secretase processing dropped. The longer the ectodomain, the less the enzyme cut. Together, these data suggested that the length of substrate ectodomain, but not its composition, matters to nicastrin.
In order to measure the substrate interaction in a controlled setting more akin to a mammalian cell, Bolduc and colleagues inserted γ-secretase and the truncated Notch-based substrate into a proteoliposome, a method previously used to study other intramembrane proteases (Dickey et al., 2013). Substrates with short ectodomains were easily cleaved by γ-secretase, but steered clear of the enzyme if they had long ectodomains.
Since nicastrin’s bulky outer portion sits atop the lipid bilayer on the extracellular side of the cell, the scientists wondered if nicastrin sterically blocked proteins with large ectodomains. A bacterial protease, RseP, works in a similar way: A large globular domain protrudes from the membrane and keeps bulky, substrate wannabes from being cleaved.
To test this, the researchers had to disrupt the nicastrin ectodomain without deleting the rest of the protein, because the γ-secretase complex cannot form without the subunit. Luckily, nicastrin is the only subunit of γ-secretase that contains disulfide bonds, and they stabilize a protein’s secondary structure. When the researchers disrupted those bridges using a reducing agent, nicastrin’s ectodomain loosened up and γ-secretase started cleaving substrates with both short and long ectodomains. The results imply that an intact ectodomain on nicastrin keeps larger substrates out of the catalytic site. This likely explains why α- or β-cleavage is required before γ-secretase can do its job: They must first chop off a good chunk of the substrate’s ectodomain so it can get past nicastrin.
The results jibe with recent high-resolution cryo-EM structures of nicastrin that place its ectodomain perpendicular to the transmembrane portion of γ-secretase, directly over the binding pocket of presenilin (see image above). The data also fit with a previous study that suggested γ-secretase preferred proteins with short ectodomains (Funamoto et al., 2013).
Bend and Flex.
The short ectodomain of a γ-secretase substrate (purple) bends underneath nicastrin (green) as it interacts with presenilin (cyan). [Courtesy of Sjors Scheres,MRC.]
What do these data mean for Alzheimer’s disease? They may explain why the APP fragment resulting from α-cleavage, C83, is a better substrate for γ-secretase than the β-cleaved product, C99. The C83 ectodomain is shorter by 16 amino acids. The data also suggest that antibodies and peptides target the N-terminal tip of C99 block γ-secretase action by bulking up the substrate’s ectodomain, which is then kept at bay by nicastrin.
“The results are impressive,” wrote Satoru Funamoto, Doshisha University, Kyoto, Japan. He noted that the shape of the substrate ectodomain—tight and compact versus loose and uncondensed—might matter as much as its length. He also pointed out that presenilin likely further selects substrates for cleavage. Bolduc agreed, suggesting that blockage by nicastrin is just the first step in γ-secretase selection. He and colleagues plan to work out how presenilin next recognizes, binds, and cuts the transmembrane domain of APP. That might help scientists design more effective therapies for Alzheimer’s disease, he told Alzforum. “If we’re ever to develop therapeutics that target Aβ production, we need to know how the peptide is made.” It may also help them understand how familial AD mutations in presenilin contribute to disease.
The data look strong, said Yueming Li, Memorial Sloan Kettering Cancer Center, New York, noting that the authors included elegant supporting kinetic and biochemical analyses. However, he cautioned that the authors only examined one of nicastrin’s substrates and wondered if this steric hindrance model applied to all of them. He said more evidence is required before scientists can definitively nail down nicastrin’s mode of action. He suggested that a high-resolution structure of a substrate in the γ-secretase active site might shed further light. A cryo EM structure published by Sjors Scheres, MRC Laboratory of Molecular Biology, Cambridge, U.K., and Yigong Shi, Tsinghua University, Beijing, in the December 1 eLIFE reveals the enzyme bound to an unknown substrate, and finds no evidence that substrates bind nicastrin (Bai et al., 2015). Rather, the substrate ectodomain slides underneath the γ-secretase subunit (see image above). That structure is consistent with the new data, said Bolduc. It implies that the substrate ectodomain has to pass under nicastrin in order to be cleaved by γ-secretase—a feat that would impossible for bulky ectodomain proteins.—Gwyneth Dickey Zakaib
References
News Citations
Paper Citations
- Shah S, Lee SF, Tabuchi K, Hao YH, Yu C, LaPlant Q, Ball H, Dann CE, Südhof T, Yu G. Nicastrin functions as a gamma-secretase-substrate receptor. Cell. 2005 Aug 12;122(3):435-47. PubMed.
- Dickey SW, Baker RP, Cho S, Urban S. Proteolysis inside the membrane is a rate-governed reaction not driven by substrate affinity. Cell. 2013 Dec 5;155(6):1270-81. PubMed.
- Funamoto S, Sasaki T, Ishihara S, Nobuhara M, Nakano M, Watanabe-Takahashi M, Saito T, Kakuda N, Miyasaka T, Nishikawa K, Saido TC, Ihara Y. Substrate ectodomain is critical for substrate preference and inhibition of γ-secretase. Nat Commun. 2013;4:2529. PubMed.
- Bai XC, Rajendra E, Yang G, Shi Y, Scheres SH. Sampling the conformational space of the catalytic subunit of human γ-secretase. Elife. 2015 Dec 1;4 PubMed.
Further Reading
Primary Papers
- Bolduc DM, Montagna DR, Gu Y, Selkoe DJ, Wolfe MS. Nicastrin functions to sterically hinder γ-secretase-substrate interactions driven by substrate transmembrane domain. Proc Natl Acad Sci U S A. 2016 Feb 2;113(5):E509-18. Epub 2015 Dec 22 PubMed.
Annotate
To make an annotation you must Login or Register.
Comments
University of Texas Southwestern Medical Center
A decade ago, we proposed a substrate receptor role for nicastrin (Shah et al., 2005). It is gratifying to see that this model has since been directly and indirectly tested and retested by a number of labs. Science can only progress when hypotheses are refuted, and models are refined and stand the test of time. In this excellent study, Bolduc and colleagues provided the latest test of the substrate receptor model, and concluded that nicastrin indeed serves as a substrate receptor for Notch, albeit only passively. Here I wish to take this opportunity to raise the following points in the hope that they will stimulate further discussion and clarify some longstanding misconceptions in the field.
First, we made two key predictions of the substrate receptor model in our studies (Shah et al., 2005; Dries et al., 2009): a) the ectodomain of nicastrin contains a substrate-binding pocket; b) the free N-terminal stubs of the immediate substrates of γ-secretase are critical for substrate recognition. More recent biochemical, functional, and structural studies from several groups have provided strong support for both of these predictions (Bai et al., 2013; Lu et al., 2014; Xie et al., 2014; Zhang et al., 2012; Hayashi et al., 2012; Funamoto et al., 2013). On the other hand, two earlier studies questioned the involvement of nicastrin in substrate recruitment (Zhao et al., 2010; Chavez-Gutierrez et al., 2008). The Bolduc study, which clearly supports the role of nicastrin in substrate gatekeeping, should perhaps help put these questions to rest and move the discussion to the precise mechanism by which nicastrin regulates substrate recognition.…More
Second, our substrate receptor model has both an active and a passive component in substrate recognition, wherein nicastrin serves as a substrate sensor and size-exclusion filter, providing an active gateway for substrates with correct N-terminal sizes to enter. The fact that the size, not the composition, of substrate N-termini determines substrate selection (Struhl et al., 2000) was a foundation of the Shah paper, and it made sense since our original discovery that the large extracellular domain of nicastrin could block the entry of APP and other protein substrates containing large amino-terminal domains (Schenk 2000, Figure 1; Yu et al., 2000). In this context, the nicastrin ectodomain and substrate N-terminal interaction provides a sensing or guiding mechanism, rather than the sole driving force recruiting the substrate for catalysis. Moreover, it is a misconception that the extracellular nicastrin-substrate interaction (or a single E333 residue alone) is the only and permanent determinant in substrate recruitment and catalysis—the interaction between substrate and γ-secretase transmembrane regions (TMRs) has also been accommodated in our original model (the substrate docking process; Shah et al., 2005, Fig7D step 4).
In our original model we noted steric compatibility for substrates (and thus hindrance to non-substrates) and the importance of substrate “docking” in the TMRs during the regulated intramembrane proteolysis (RIP) cycle. We stated that “Considering that nicastrin and γ-secretase substrates are anchored in a two-dimensional lipid bilayer by their TMRs, the interaction of the short extracellular N termini of type I membrane proteins with the nicastrin ectodomain is constrained and depends on the spatial distance and steric compatibility of their respective binding sites … The captured substrates are aligned and docked in the lipid bilayer, likely at the substrate-docking site in the TMRs of presenilin and presented to the catalytic core for intramembrane cleavage” (Shah et al., 2005, Page 445, third paragraph and Figure 7D). Thus, the observation by Bolduc and colleagues that nicastrin plays a steric blocking role is consistent with the receptor model, and the differences between the two models are somewhat semantic. At its core, both the passive receptor model advanced by Bolduc and colleagues and the receptor model advanced by us emphasize the importance of the nicastrin ectodomain and the size of the N-termini of substrates, and in the Bolduc model, the N-termini of substrates might still need to be accommodated by nicastrin.
Third, while both models suggest that nicastrin is a substrate receptor, it is implausible that nicastrin only plays a purely passive steric hindrance role in substrate recognition. There are many physiologically important type-I membrane proteins with naturally short N-termini that are not generated by ectodomain shedding. It would be problematic if these normal proteins were processed uncontrollably in vivo. Entry of type-II membrane proteins with short C-termini could also be a problem if nicastrin only passively regulates substrate entry via steric hindrance. Contrary to the views of Bolduc and colleagues, the evidences presented in their study are not incompatible with an active role for nicastrin in substrate recruitment. Rather, it seems that some of the main differences between the Bolduc model and the Shah model are based on differing interpretations from similar experiments.
For example, we had showed that an APP-derived TMR peptide could be processed in vitro without involvement of the substrate-recognition process associated with the nicastrin ectodomain (Shah et al., 2005, Fig5E). Similar experiments by Bolduc and colleagues also yielded results that substrates could be made to bypass the nicastrin ectodomain in vitro, but these results were interpreted as arguing against an active role for nicastrin. However, our initial model predicted this outcome: Our model requires nicastrin to sense the substrate ectodomain, so if such a domain does not exist or is compromised, then the substrate will subsequently bypass nicastrin and be cleaved at the catalytic core, at least in vitro.
Bolduc and colleagues considered that the observation that relaxing the structural fold of nicastrin ectodomain allows for more efficient cleavage of substrate retaining longer N-terminal stubs is evidence against an active role for nicastrin in substrate recognition. We respectfully disagree with such interpretation and believe this observation is compatible with the substrate receptor model and only indicates that the nicastrin ectodomain is involved in substrate selection. It is entirely logical for the catalytic core of an enzyme to be active when a regulatory domain is removed or compromised, but this does not mean the regulatory domain does not exist or is not important. Nicastrin is the gatekeeper to the catalytic site, and when this gatekeeper is compromised, the entry of substrate into the catalytic core should be easier, particularly for uninvited guests.
The high-resolution structures of nicastrin and γ-secretase showed that the nicastrin substrate-binding site and the presenilin catalytic site are on opposite sides of the γ-secretase complex, and Bolduc and colleagues cited this as other evidence against an active role for nicastrin. However, this arrangement was clearly predicted in our original model (Shah et al., 2005, Figure 7D, steps 3-4) and cannot be used as evidence to disprove that nicastrin plays an active role in substrate recruitment, just as misalignment of the two aspartate residues of presenilin (in its inactive state) (Bai et al., 2013) do not argue against a role of these residues in catalysis (in its active state). As a matter of fact, a conformational change (perhaps induced by high affinity docking of the substrate TMR to γ-secretase) could easily provide a satisfactory explanation for controlled substrate access and realignment of the nicastrin substrate-binding site with the presenilin catalytic site, and there is good evidence that such conformational change exists in γ-secretase (Li et al., 2014). It is also premature to make a conclusion when the nature of the “unknown substrate” is unknown and the conformational state of the “Bend and Flex” complex (see above) in the RIP cycle is unclear. Regardless of the speculative differences in mechanistic details, it is satisfying to see that our original studies have advanced the thinking and practice of the field over the past decade and that Bolduc and colleagues now provided additional evidence that nicastrin plays a role in substrate recognition, whether passively, actively, or more likely a combination of both. Clearly, more work is needed to further uncover the details of the mechanistic action of nicastrin in substrate recruitment during the entire RIP cycle and to refine the substrate receptor model of nicastrin when justified by new evidences.
References:
Shah S, Lee SF, Tabuchi K, Hao YH, Yu C, LaPlant Q, Ball H, Dann CE, Südhof T, Yu G. Nicastrin functions as a gamma-secretase-substrate receptor. Cell. 2005 Aug 12;122(3):435-47. PubMed.
Dries DR, Shah S, Han YH, Yu C, Yu S, Shearman MS, Yu G. Glu-333 of nicastrin directly participates in gamma-secretase activity. J Biol Chem. 2009 Oct 23;284(43):29714-24. PubMed.
Bai XC, Yan C, Yang G, Lu P, Ma D, Sun L, Zhou R, Scheres SH, Shi Y. An atomic structure of human γ-secretase. Nature. 2015 Sep 10;525(7568):212-7. Epub 2015 Aug 17 PubMed.
Lu P, Bai XC, Ma D, Xie T, Yan C, Sun L, Yang G, Zhao Y, Zhou R, Scheres SH, Shi Y. Three-dimensional structure of human γ-secretase. Nature. 2014 Aug 14;512(7513):166-70. Epub 2014 Jun 29 PubMed.
Xie T, Yan C, Zhou R, Zhao Y, Sun L, Yang G, Lu P, Ma D, Shi Y. Crystal structure of the γ-secretase component nicastrin. Proc Natl Acad Sci U S A. 2014 Sep 16;111(37):13349-54. Epub 2014 Sep 2 PubMed.
Zhang X, Hoey RJ, Lin G, Koide A, Leung B, Ahn K, Dolios G, Paduch M, Ikeuchi T, Wang R, Li YM, Koide S, Sisodia SS. Identification of a tetratricopeptide repeat-like domain in the nicastrin subunit of γ-secretase using synthetic antibodies. Proc Natl Acad Sci U S A. 2012 May 29;109(22):8534-9. PubMed.
Hayashi I, Takatori S, Urano Y, Miyake Y, Takagi J, Sakata-Yanagimoto M, Iwanari H, Osawa S, Morohashi Y, Li T, Wong PC, Chiba S, Kodama T, Hamakubo T, Tomita T, Iwatsubo T. Neutralization of the γ-secretase activity by monoclonal antibody against extracellular domain of nicastrin. Oncogene. 2011 Jul 4; PubMed.
Funamoto S, Sasaki T, Ishihara S, Nobuhara M, Nakano M, Watanabe-Takahashi M, Saito T, Kakuda N, Miyasaka T, Nishikawa K, Saido TC, Ihara Y. Substrate ectodomain is critical for substrate preference and inhibition of γ-secretase. Nat Commun. 2013;4:2529. PubMed.
Zhao G, Liu Z, Ilagan MX, Kopan R. Gamma-secretase composed of PS1/Pen2/Aph1a can cleave notch and amyloid precursor protein in the absence of nicastrin. J Neurosci. 2010 Feb 3;30(5):1648-56. PubMed.
Chávez-Gutiérrez L, Tolia A, Maes E, Li T, Wong PC, De Strooper B. Glu(332) in the Nicastrin ectodomain is essential for gamma-secretase complex maturation but not for its activity. J Biol Chem. 2008 Jul 18;283(29):20096-105. PubMed.
Struhl G, Adachi A. Requirements for presenilin-dependent cleavage of notch and other transmembrane proteins. Mol Cell. 2000 Sep;6(3):625-36. PubMed.
Schenk D. Alzheimer's disease. A partner for presenilin. Nature. 2000 Sep 7;407(6800):34-5. PubMed.
Yu G, Nishimura M, Arawaka S, Levitan D, Zhang L, Tandon A, Song YQ, Rogaeva E, Chen F, Kawarai T, Supala A, Levesque L, Yu H, Yang DS, Holmes E, Milman P, Liang Y, Zhang DM, Xu DH, Sato C, Rogaev E, Smith M, Janus C, Zhang Y, Aebersold R, Farrer LS, Sorbi S, Bruni A, Fraser P, St George-Hyslop P. Nicastrin modulates presenilin-mediated notch/glp-1 signal transduction and betaAPP processing. Nature. 2000 Sep 7;407(6800):48-54. PubMed.
Li Y, Lu SH, Tsai CJ, Bohm C, Qamar S, Dodd RB, Meadows W, Jeon A, McLeod A, Chen F, Arimon M, Berezovska O, Hyman BT, Tomita T, Iwatsubo T, Johnson CM, Farrer LA, Schmitt-Ulms G, Fraser PE, St George-Hyslop PH. Structural interactions between inhibitor and substrate docking sites give insight into mechanisms of human PS1 complexes. Structure. 2014 Jan 7;22(1):125-35. Epub 2013 Nov 7 PubMed.
The University of Tokyo
I found this paper quite intriguing, as it reveals a novel aspect of the function of nicastrin in the proteolytic activity of γ-secretase. Also, their results provide a novel mechanistic interpretation for anti-nicastrin extracellular domain (ECD) antibodies that have inhibitory activity against γ-secretase, suggesting additional steric hindrance by antibodies against substrate capture (Hayashi et al.,. 2011; Zhang et al., 2012).
However, these results shed no light on the "biological" function of nicastrin in the maturation/trafficking of the γ-secretase complex. Genetically, it is apparent that nicastrin is required for the complex to form, though what role nicastrin plays remains unresolved. Also, it would be intriguing to test the activity of ECD-less nicastrin on γ-secretase in the living cell. This might be important for understanding the molecular mechanism of acne inversa/hidradenitis suppurativa, in which loss-of-function mutations in NCSTN lead to haploinsufficiency. …More
Doshisha University
This study by the groups of Selkoe and Wolfe adds further details about functional role of nicastrin in the γ-secretase complex. The most prominent finding is that nicastrin is a gatekeeper rather than a substrate receptor in the complex (Shah et al., 2005). The results are impressive, showing increased cleavage efficiency of this enzyme after disruption of nicastrin folds in the presence of reducing reagents. Their data provide insights into why substrates with large ectodomains fail to be cleaved by this enzyme (Struhl and Adachi, 2000). This finding is consistent with our previous report that γ-secretase prefers substrates with shorter ectodomains and that our C99-binding peptide causes steric hindrance between C99 and γ-secretase complex (Funamoto et al., 2013). Although γ-secretase preferentially cleaves substrates with shorter ectodomains, it is unlikely that the substrate ectodomain is a straight protrusion from the transmembrane domain. Presumably, the compactness of the structure, rather than polypeptide length, is critical to recognition by γ-secretase. …More
The authors showed that complete deletion of ectodomain increased the Km value, which suggests that substrate transmembrane domain forced the interaction with γ-secretase. We observed that a C99 substrate carrying the Notch transmembrane domain tended to increase interaction with γ-secretase components and cleavage efficiency (Funamoto et al., 2013). These findings suggest that the composition of the transmembrane domain of the substrate is also involved in the substrate specificity of γ-secretase. As the authors pointed out, it will be critical to define the nature of the interaction between substrate transmembrane domain and γ-secretase active site.
References:
Struhl G, Adachi A. Requirements for presenilin-dependent cleavage of notch and other transmembrane proteins. Mol Cell. 2000 Sep;6(3):625-36. PubMed.
Shah S, Lee SF, Tabuchi K, Hao YH, Yu C, LaPlant Q, Ball H, Dann CE, Südhof T, Yu G. Nicastrin functions as a gamma-secretase-substrate receptor. Cell. 2005 Aug 12;122(3):435-47. PubMed.
Funamoto S, Sasaki T, Ishihara S, Nobuhara M, Nakano M, Watanabe-Takahashi M, Saito T, Kakuda N, Miyasaka T, Nishikawa K, Saido TC, Ihara Y. Substrate ectodomain is critical for substrate preference and inhibition of γ-secretase. Nat Commun. 2013;4:2529. PubMed.
Make a Comment
To make a comment you must login or register.