A New Villain: Nitrated Aβ May Seed Plaques, Damage Memory
Quick Links
In the search for what makes Aβ turn rogue and start gumming up the brain, a paper in the September 8 Neuron fingers a novel suspect. Researchers led by Michael Heneka at the University of Bonn, Germany, report that modifying human Aβ by adding a nitro group enhanced the peptide’s tendency to aggregate. Heneka and colleagues saw nitrated Aβ at the core of amyloid plaques in both human and mouse Alzheimer’s brains. In addition, nitrated Aβ was able to seed plaques in AD model mice, whereas the non-nitrated form could not. Regardless of its aggregation state, nitrated Aβ also damaged synaptic function and memory in mice. When the researchers blocked a nitration pathway in these animals, the plaque load dropped and learning ability returned to normal. The results suggest a potential new pathway for intervention in AD pathology, although it remains to be seen if the results will hold up across labs and translate to people.
The findings further link inflammation to amyloid pathology. Inflammation has been shown to turn on the production of nitric oxide synthase 2 (NOS2) in neurons, astrocytes, and microglia (see, e.g., Vodovotz et al., 1996; Heneka et al., 2001; and Fernández-Vizarra et al., 2004). NOS2 then catalyzes the formation of nitric oxide (NO), which leads to protein nitration. With the new finding that nitrated Aβ can seed plaques, this suggests that inflammation is not merely a side show in AD, but can “accelerate and drive the degenerative pathology,” Heneka told ARF. Intriguingly, a recent study showed that Aβ aggregates can increase the formation of NO (see Du et al., 2011). In addition, amyloid plaques are known to stimulate glial cells, leading to more NOS2 production, in what could be a vicious cycle that might help spread the disease through the brain, Heneka suggested.
Nitrated proteins are common in AD brains (see, e.g., Hensley et al., 1998; ARF related news story on Lüth et al., 2002; and Castegna et al., 2003). They may play a role in synaptic injury and cell death (see Radi, 2004 and Nakamura and Lipton, 2011). Nitrated tau has been linked to tangle formation (Horiguchi et al., 2003), and α-synuclein aggregates are heavily nitrated (see ARF related news story on Giasson et al., 2000). However, no one had looked closely at Aβ nitration.
To investigate this, first author Markus Kummer showed that Aβ can be nitrated at tyrosine residue 10 in vitro, and that nitrated Aβ clumps together more readily than the normal form does. Kummer and colleagues developed an antibody that specifically recognizes the nitrated form of Aβ. In both human brains and in APP/PS1 transgenic mice, the antibody labeled the core of amyloid plaques. In mouse brain, the nitrated core did not grow in size over several months, but non-nitrated Aβ continued to glom onto the plaque, suggesting the nitrated form might act to seed plaques. In support of this, when the researchers injected either nitrated or non-nitrated Aβ into mouse brains and looked eight weeks later, only the mice that had received nitrated Aβ had numerous small plaques scattered through their brain tissue.
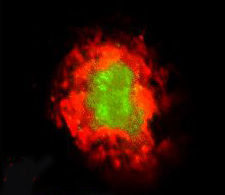
Nitrated Aβ (green) sits at the core of amyloid plaques (red) in transgenic mouse brain. Image cedit: Michael Heneka
Other evidence suggests a key role for nitration in kicking off amyloid aggregation, Heneka told ARF. “All mammals who develop Aβ plaque deposition carry a tyrosine at position 10,” Heneka said, citing primates, chickens, and guinea pigs as examples. Mice and other mammals that do not get plaques have a different amino acid at that position and their Aβ cannot be nitrated, Heneka said. Likewise, synthetic, unmodified Aβ does not initiate amyloidosis in brain, but Aβ from brain extracts does (see ARF related news story on Eisele et al., 2009). In future work, Heneka would like to generate a transgenic mouse that carries rodent Aβ modified to have a tyrosine at position 10, and see if these animals develop amyloid deposits.
Kummer and colleagues also looked at what happened when they reduced protein nitration by knocking out or enzymatically inhibiting NOS2. APP/PS1 mice without NOS2 had a lower plaque load than normal APP/PS1 mice, and their spatial memory deficits disappeared. Memory improved even in young AD mice that did not yet have plaques, Heneka told ARF. The protective effect may be mediated through synapses, the authors suggest, as blocking NOS2 also rescued long-term potentiation (LTP) in both young and old APP/PS1 mice. In wild-type hippocampal slices, nitrated Aβ reduced LTP more than regular Aβ did.
The results agree with a previous study led by Flint Beal at Weill Cornell Medical College, New York City, that found less AD pathology in crossbred Tg2576 and PS1 mice when they were crossed with NOS2 knockouts (see Nathan et al., 2005). However, the findings contrast with studies by Carol Colton and Mike Vitek at Duke University, Durham, North Carolina, and David Wink at the National Cancer Institute, Bethesda, Maryland, who reported that knocking out NOS2 worsens Aβ pathology and memory in several mutant APP mouse models (see ARF related news story on Colton et al., 2006; ARF related news story on Wilcock et al., 2008; and Colton et al., 2008). The main difference between the models used by these groups is the presence of mutant presenilin, which points to it as the reason for the differential response to the nitric oxide environment, Colton and colleagues wrote to ARF (see full comment below). This is important because mutated presenilin is present in less than 1 percent of human AD cases. Colton and colleagues also point out that NOS2 and NO have diverse biological effects, many of them beneficial for cell growth and survival, and that nitration can occur through routes other than NOS2, raising the question of whether NOS2 inhibition is the best route for preventing Aβ nitration.
However, Heneka notes that a previous study found that NOS2 deletion in wild-type mouse hippocampal slices protected synapses from Aβ-mediated damage (see ARF related news story on Wang et al., 2004), suggesting the strategy could be beneficial even in the absence of presenilin mutations. The NOS2 inhibitor that Heneka’s group used, L-NIL, has been tested in humans as a possible anti-asthmatic (see Hansel et al., 2003), and shown to enter the mouse brain (see Rebello et al., 2002). Heneka would like to do human trials, but has no funding yet. Meanwhile, he is testing vaccination against nitrated Aβ in mice. Current human vaccination approaches may not clear nitrated Aβ, Heneka believes, speculating that the absence, to date, of reported cognitive improvement despite evidence of brain Aβ removal might be due to the continued presence of this newly proposed villain.—Madolyn Bowman Rogers
References
News Citations
- No Junk After All: Dinucleotide Repeats Change eNOS Splicing; What about AD?
- Linking Free Radicals to Neurodegenerative Disease
- Aβ the Bad Apple? Seeding and Propagating Amyloidosis
- No NOS Promotes Tau Pathology in APP Transgenic Mice
- NOS Knockout Unleashes AD Pathology, Neuronal Death in CAA Mice
- Microglia Aid and Abet Aβ Toxicity and Clearance
Paper Citations
- Vodovotz Y, Lucia MS, Flanders KC, Chesler L, Xie QW, Smith TW, Weidner J, Mumford R, Webber R, Nathan C, Roberts AB, Lippa CF, Sporn MB. Inducible nitric oxide synthase in tangle-bearing neurons of patients with Alzheimer's disease. J Exp Med. 1996 Oct 1;184(4):1425-33. PubMed.
- Heneka MT, Wiesinger H, Dumitrescu-Ozimek L, Riederer P, Feinstein DL, Klockgether T. Neuronal and glial coexpression of argininosuccinate synthetase and inducible nitric oxide synthase in Alzheimer disease. J Neuropathol Exp Neurol. 2001 Sep;60(9):906-16. PubMed.
- Fernández-Vizarra P, Fernández AP, Castro-Blanco S, Encinas JM, Serrano J, Bentura ML, Muñoz P, Martínez-Murillo R, Rodrigo J. Expression of nitric oxide system in clinically evaluated cases of Alzheimer's disease. Neurobiol Dis. 2004 Mar;15(2):287-305. PubMed.
- Du XT, Wang L, Wang YJ, Andreasen M, Zhan DW, Feng Y, Li M, Zhao M, Otzen D, Xue D, Yang Y, Liu RT. Aβ1-16 can aggregate and induce the production of reactive oxygen species, nitric oxide, and inflammatory cytokines. J Alzheimers Dis. 2011;27(2):401-13. PubMed.
- Hensley K, Maidt ML, Yu Z, Sang H, Markesbery WR, Floyd RA. Electrochemical analysis of protein nitrotyrosine and dityrosine in the Alzheimer brain indicates region-specific accumulation. J Neurosci. 1998 Oct 15;18(20):8126-32. PubMed.
- Lüth HJ, Münch G, Arendt T. Aberrant expression of NOS isoforms in Alzheimer's disease is structurally related to nitrotyrosine formation. Brain Res. 2002 Oct 25;953(1-2):135-43. PubMed.
- Castegna A, Thongboonkerd V, Klein JB, Lynn B, Markesbery WR, Butterfield DA. Proteomic identification of nitrated proteins in Alzheimer's disease brain. J Neurochem. 2003 Jun;85(6):1394-401. PubMed.
- Radi R. Nitric oxide, oxidants, and protein tyrosine nitration. Proc Natl Acad Sci U S A. 2004 Mar 23;101(12):4003-8. PubMed.
- Nakamura T, Lipton SA. Redox modulation by S-nitrosylation contributes to protein misfolding, mitochondrial dynamics, and neuronal synaptic damage in neurodegenerative diseases. Cell Death Differ. 2011 Sep;18(9):1478-86. PubMed.
- Horiguchi T, Uryu K, Giasson BI, Ischiropoulos H, LightFoot R, Bellmann C, Richter-Landsberg C, Lee VM, Trojanowski JQ. Nitration of tau protein is linked to neurodegeneration in tauopathies. Am J Pathol. 2003 Sep;163(3):1021-31. PubMed.
- Giasson BI, Duda JE, Murray IV, Chen Q, Souza JM, Hurtig HI, Ischiropoulos H, Trojanowski JQ, Lee VM. Oxidative damage linked to neurodegeneration by selective alpha-synuclein nitration in synucleinopathy lesions. Science. 2000 Nov 3;290(5493):985-9. PubMed.
- Eisele YS, Bolmont T, Heikenwalder M, Langer F, Jacobson LH, Yan ZX, Roth K, Aguzzi A, Staufenbiel M, Walker LC, Jucker M. Induction of cerebral beta-amyloidosis: intracerebral versus systemic Abeta inoculation. Proc Natl Acad Sci U S A. 2009 Aug 4;106(31):12926-31. PubMed.
- Nathan C, Calingasan N, Nezezon J, Ding A, Lucia MS, La Perle K, Fuortes M, Lin M, Ehrt S, Kwon NS, Chen J, Vodovotz Y, Kipiani K, Beal MF. Protection from Alzheimer's-like disease in the mouse by genetic ablation of inducible nitric oxide synthase. J Exp Med. 2005 Nov 7;202(9):1163-9. PubMed.
- Colton CA, Vitek MP, Wink DA, Xu Q, Cantillana V, Previti ML, Van Nostrand WE, Weinberg JB, Weinberg B, Dawson H. NO synthase 2 (NOS2) deletion promotes multiple pathologies in a mouse model of Alzheimer's disease. Proc Natl Acad Sci U S A. 2006 Aug 22;103(34):12867-72. PubMed.
- Wilcock DM, Lewis MR, Van Nostrand WE, Davis J, Previti ML, Gharkholonarehe N, Vitek MP, Colton CA. Progression of amyloid pathology to Alzheimer's disease pathology in an amyloid precursor protein transgenic mouse model by removal of nitric oxide synthase 2. J Neurosci. 2008 Feb 13;28(7):1537-45. PubMed.
- Colton CA, Wilcock DM, Wink DA, Davis J, Van Nostrand WE, Vitek MP. The effects of NOS2 gene deletion on mice expressing mutated human AbetaPP. J Alzheimers Dis. 2008 Dec;15(4):571-87. PubMed.
- Wang Q, Rowan MJ, Anwyl R. Beta-amyloid-mediated inhibition of NMDA receptor-dependent long-term potentiation induction involves activation of microglia and stimulation of inducible nitric oxide synthase and superoxide. J Neurosci. 2004 Jul 7;24(27):6049-56. PubMed.
- Hansel TT, Kharitonov SA, Donnelly LE, Erin EM, Currie MG, Moore WM, Manning PT, Recker DP, Barnes PJ. A selective inhibitor of inducible nitric oxide synthase inhibits exhaled breath nitric oxide in healthy volunteers and asthmatics. FASEB J. 2003 Jul;17(10):1298-300. PubMed.
Other Citations
External Citations
Further Reading
News
- Microglia Aid and Abet Aβ Toxicity and Clearance
- NOS Knockout Unleashes AD Pathology, Neuronal Death in CAA Mice
- Aβ the Bad Apple? Seeding and Propagating Amyloidosis
- No Junk After All: Dinucleotide Repeats Change eNOS Splicing; What about AD?
- Linking Free Radicals to Neurodegenerative Disease
- No NOS Promotes Tau Pathology in APP Transgenic Mice
Primary Papers
- Kummer MP, Hermes M, Delekarte A, Hammerschmidt T, Kumar S, Terwel D, Walter J, Pape HC, König S, Roeber S, Jessen F, Klockgether T, Korte M, Heneka MT. Nitration of tyrosine 10 critically enhances amyloid β aggregation and plaque formation. Neuron. 2011 Sep 8;71(5):833-44. PubMed.
Annotate
To make an annotation you must Login or Register.
Comments
Duke University Medical Center and Cognosci
The Case for Personalized Alzheimer’s Medicine
Heneka and colleagues’ new report on the involvement of nitric oxide (NO) in Alzheimer’s disease (AD) serves to further underscore the need for properly matching the treatment to the unique patient. They report that APP/PS1 transgenic mice develop robust amyloid plaque pathology containing amyloid-β peptide (Aβ) that is modified by nitration of tyrosine at position 10. As did previous groups (Smith et al., 1997), they also find 3-nitrotyrosine-modified (3-NTyr) Aβ in the brains of AD patients and suggest that modifications to Aβ can seed or stimulate plaque formation. Similar to our earlier reports (Vitek et al., 1994; Smith et al., 1995), they also find that post-translational modifications of Aβ appear to occur at relatively younger ages when pathology is thought to begin developing. Interestingly, while deposited Aβ does appear to increase with time, the amount of nitrotyrosine-Aβ does not increase over time in this model. These data provoke the question of how nitric oxide, nitrative stress, and Aβ interact over the course of the disease.
At this point, there are simply many critical questions with no unambiguous answers. For example, it is not even clear if brain iNOS activity is meaningfully increased, particularly in humans, or if NO production is maintained in AD, if NO is, in fact, the sole product of iNOS activation. Coupled with the lack of arginine due to increased arginase 1 expression, NOS actually generates superoxide anion. It is not clear if iNOS is increased only at certain disease stages, or if the levels of NO generated by iNOS in vivo are sufficient to have the modifying effects demonstrated by direct application of nitrating/oxidizing agents to a protein. Furthermore, nitration of amino acids does not require enzymatically generated NO. It is now well known that 3-nitrotyrosine adducts can be formed by nitrite, hydrogen peroxide, and free metals, as well as by mechanisms that incorporate peroxidase activity (Thomas et al., 2002; Thomas et al., 2006). In AD or in mouse models of AD, the accumulation of Aβ fibrils that are also well known to bind to and produce a surface for reactive metals such as iron and copper (Dikalov et al., 2004) are highly likely to facilitate non-enzymatic nitration processes. That the 3-NTyr Aβ residue was localized to the core of the plaques in the present study further supports this possibility. In fact, because NO can be rapidly scavenged by superoxide anion, Aβ-mediated reactions are a mechanism by which bioavailability of NO is reduced and critical growth supporting/survival mechanisms mediated by NO are compromised. This loss of NO may account for the failure to observe an increase in 3-NTyr Aβ at the nine-month time point as shown in Figure 2.
With strong evidence that 3-NTyr-Aβ can stimulate aggregation and deposition of plaques, the experiments turn to understanding the consequences of removing nitric oxide from these APP/PS1 mice. Using both genetic knockout of the NOS2 gene, and the L-NIL inhibitor of iNOS, they find decreased 3-NTyr-Aβ, decreased Aβ deposits, and improved performance on a radial arm maze behavioral task with no change in tau pathology or neuronal loss reported. These findings are in stark contrast to our publications where APP transgenic mice on a NOS2 knockout background (Tg2576/NOS2 knockout or APP-SwDI/NOS2 knockout and, most recently, the 5xFAD/NOS2 knockout) develop robust plaque pathology, phospho-tau deposits, neuronal loss, and behavioral deficits (Colton et al., 2006; Wilcock et al., 2008; Colton et al., 2008). We’d like to note what we consider an inaccuracy in the paper’s discussion on this point. First, we do not see improved behavioral effects in our mouse models; rather, we see severe behavioral impairment with APP/NOS2-/-. Second, the concept of altered microglia was simply one of multiple possibilities given to explain the variance in data between Nathan et al., 2005, and our own. Similar to Nathan’s report that amyloid pathology of an APP/PS1 mouse was significantly reduced when placed on a NOS2 knockout background, the main difference between Heneka’s and our work is the presence of a mutated presenilin-1 gene. Given the contrasting findings, we conclude that the mutated PS1 gene is responsible for a differential response to the nitric oxide environment.
Mutated PS1 genes are found in less than 1 percent of all Alzheimer’s patients. Heneka and Nathan’s work suggests, importantly, that inhibitors of nitric oxide synthase may be appropriate for these patients. For the other 99 percent, our work clearly shows that nitric oxide exerts a protective effect that reduces plaque and tangle burdens, reduces neuronal loss, and improves behavior. With the ability to rapidly diagnose the presence of non-mutated and mutated PS1 genes in suspected AD patients, the field is poised to directly test the risks and benefits of NOS inhibitors and nitric oxide donors as potential therapies for this devastating disease.
References:
Smith MA, Richey Harris PL, Sayre LM, Beckman JS, Perry G. Widespread peroxynitrite-mediated damage in Alzheimer's disease. J Neurosci. 1997 Apr 15;17(8):2653-7. PubMed.
Vitek MP, Bhattacharya K, Glendening JM, Stopa E, Vlassara H, Bucala R, Manogue K, Cerami A. Advanced glycation end products contribute to amyloidosis in Alzheimer disease. Proc Natl Acad Sci U S A. 1994 May 24;91(11):4766-70. PubMed.
Smith MA, Sayre LM, Vitek MP, Monnier VM, Perry G. Early AGEing and Alzheimer's. Nature. 1995 Mar 23;374(6520):316. PubMed.
Thomas DD, Espey MG, Vitek MP, Miranda KM, Wink DA. Protein nitration is mediated by heme and free metals through Fenton-type chemistry: an alternative to the NO/O2- reaction. Proc Natl Acad Sci U S A. 2002 Oct 1;99(20):12691-6. PubMed.
Thomas DD, Espey MG, Pociask DA, Ridnour LA, Donzelli S, Wink DA. Asbestos redirects nitric oxide signaling through rapid catalytic conversion to nitrite. Cancer Res. 2006 Dec 15;66(24):11600-4. PubMed.
Dikalov SI, Vitek MP, Mason RP. Cupric-amyloid beta peptide complex stimulates oxidation of ascorbate and generation of hydroxyl radical. Free Radic Biol Med. 2004 Feb 1;36(3):340-7. PubMed.
Colton CA, Vitek MP, Wink DA, Xu Q, Cantillana V, Previti ML, Van Nostrand WE, Weinberg JB, Weinberg B, Dawson H. NO synthase 2 (NOS2) deletion promotes multiple pathologies in a mouse model of Alzheimer's disease. Proc Natl Acad Sci U S A. 2006 Aug 22;103(34):12867-72. PubMed.
Wilcock DM, Lewis MR, Van Nostrand WE, Davis J, Previti ML, Gharkholonarehe N, Vitek MP, Colton CA. Progression of amyloid pathology to Alzheimer's disease pathology in an amyloid precursor protein transgenic mouse model by removal of nitric oxide synthase 2. J Neurosci. 2008 Feb 13;28(7):1537-45. PubMed.
Colton CA, Wilcock DM, Wink DA, Davis J, Van Nostrand WE, Vitek MP. The effects of NOS2 gene deletion on mice expressing mutated human AbetaPP. J Alzheimers Dis. 2008 Dec;15(4):571-87. PubMed.
Nathan C, Calingasan N, Nezezon J, Ding A, Lucia MS, La Perle K, Fuortes M, Lin M, Ehrt S, Kwon NS, Chen J, Vodovotz Y, Kipiani K, Beal MF. Protection from Alzheimer's-like disease in the mouse by genetic ablation of inducible nitric oxide synthase. J Exp Med. 2005 Nov 7;202(9):1163-9. PubMed.
View all comments by Michael VitekHertie Institute for Clinical Brain Research, University of Tübingen, and DZNE Tübingen
This is a very nice study by Heneka et al. Before readers reach conclusions about the nature of the amyloidogenic seed, I'd like to offer a note of caution. The authors used 0.25 mg/ml synthetic or synthetic nitrated Aβ. This is at least 50 times more Aβ than what is in the brain extract that we used for seeding in Meyer-Luehmann et al., 2006 (see Figure 4). Moreover, the brain extract we used induced much more amyloid induction than what is reported here; 0.25 mg/ml is 50,000 times more Aβ than what is contained in a 100,000 g supernatant of a brain, which also has significant seeding capacity (Langer et al., in press).
We also once used 0.5 mg/ml synthetic Aβ in Meyer-Luehmann et al. (supplemental Table). With that we also saw some amyloid after a four-month incubation period, but this was in large part the injected material similar to that reported in Heneka et al. However, in Heneka et al. it is very interesting to see some endogenous Aβ binding (or co-aggregating) to the injected nitrated Aβ.
Thus, in my view, from the presented results, the nitrated Aβ cannot (yet) be considered the magic seeding bullet, and more studies are necessary. But this is a very interesting paper.
References:
Meyer-Luehmann M, Coomaraswamy J, Bolmont T, Kaeser S, Schaefer C, Kilger E, Neuenschwander A, Abramowski D, Frey P, Jaton AL, Vigouret JM, Paganetti P, Walsh DM, Mathews PM, Ghiso J, Staufenbiel M, Walker LC, Jucker M. Exogenous induction of cerebral beta-amyloidogenesis is governed by agent and host. Science. 2006 Sep 22;313(5794):1781-4. PubMed.
View all comments by Mathias JuckerMake a Comment
To make a comment you must login or register.