Mutant Presenilin Skews Calcium Homeostasis by Chomping on ER Sensor
Quick Links
Researchers have unearthed a new victim of presenilin 1’s voracious appetite. According to a study published September 6 in Science Signaling, the enzyme cleaves STIM1—a calcium sensor in the endoplasmic reticulum that promotes the replenishment of the cation when stores run low. The researchers, led by King-Ho Cheung of the University of Hong Kong, reported that familial Alzheimer’s disease (FAD) mutations in presenilin enhanced cleavage of STIM1, causing calcium influx to flag. The calcium dearth crumbled mature dendritic spines on the surface of neurons. The researchers proposed that in addition to its role in churning out Aβ peptides by processing APP, presenilin 1 also contributes to neuronal dysfunction via its destruction of STIM1.
“We have known for some time that presenilin mutations associated with FAD alter calcium signaling in neurons, but how this occurred was a mystery,” commented Stanley Thayer of the University of Minnesota Medical School in Minneapolis, who was not involved in the study. “The report by Tong et al. provides a mechanism linking the altered calcium signaling to the enzymatic activity of γ-secretase.”
Normally, neuronal stimulation triggers a release of calcium from the ER, which is then rapidly replaced by an influx of calcium through channels on the cell surface. This replenishment occurs when STIM1 and STIM2 calcium receptors in the ER sense low calcium levels and communicate with receptors on the cell surface to promote calcium influx. This so-called capacitative calcium entry (CCE), also known as store-operated calcium entry (SOCE), reportedly weakens in capacitative calcium entry cells expressing mutant presenilin (see Leissring et al., 2000; Yoo et al., 2000).
Researchers have proffered multiple explanations for this, including the idea, proposed by Ilya Bezprozvanny’s group at the University of Texas Southwestern Medical Center in Dallas, that PS1 is itself an ER calcium release channel (see Sep 2006 news; Jun 2010 news). As a postdoc in Kevin Foskett’s lab at the University of Pennsylvania in Philadelphia, Cheung published several studies supporting yet another mode of PS1 influence on calcium signaling, through an interaction with inositol triphosphate (IP3) receptors in the ER membrane that promotes ER calcium release (see Jun 2008 news on Cheung et al., 2008; Cheung et al., 2010; May 2014 news). Bezprozvanny subsequently reported that PS1 somehow lowers the expression of STIM2, thus preventing influx of calcium through that route as well (see Apr 2014 news).
First author Benjamin Chun-Kit Tong and colleagues wondered if calcium influx related to PS1’s protease activity. PS1 only gains catalytic activity as part of the larger γ-secretase complex, and despite its purported solo role as a calcium leak channel, earlier studies indicated PS1 effects on calcium homeostasis partially require catalytic activity (see Akbari et al., 2004; Bojarski et al., 2009).
The researchers started by comparing CCE in neuroblastoma cells expressing either wild-type PS1 or PS1 harboring the M146L, A246E, V97L, or A136G mutations. They depleted ER calcium by stimulating the cells with an acetylcholine receptor agonist in the absence of calcium, then monitored calcium influx after adding the cation back to the medium. Using single-cell calcium imaging, they found that cells expressing any of the mutant forms of PS1 took up calcium more slowly—and took up less of it—than cells expressing normal PS1. The same was true when the researchers performed the experiments using skin fibroblasts from AD patients with PS1 mutations. Tellingly, CCE occurred at normal levels in all the cells expressing mutant forms of PS1 when the researchers pretreated the cells with DAPT, a γ-secretase inhibitor. Together, these findings indicated that FAD-linked PS1 mutations attenuated calcium replenishment, and that PS1’s catalytic activity was required for this effect.
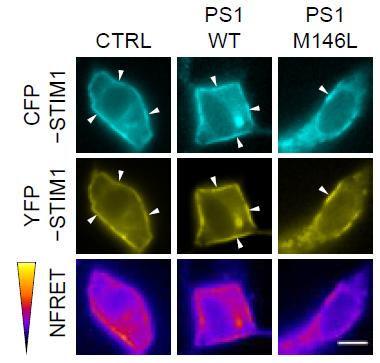
Rendezvous Ruined.
In control cells or those expressing normal PS1 (left and middle panels), STIM1 molecules come together to produce a FRET signal (magenta, bottom). This meet-up is reduced in cells expressing PS1-M146L. [Image courtesy of Tong et al., Science Signaling, 2016.]
Further experiments indicated that STIM1 and PS1 physically interacted in the ER, and that mutated forms of PS1 prevented the oligomerization of STIM1 in response to waning ER calcium. This oligomerization allows ER-bound STIM1 to move to plasma membrane junctions, where it interacts with the ORAI-1 receptor, which ultimately triggers calcium influx through the plasma membrane. Using a combination of fluorescence resonance energy transfer (FRET) and total internal reflection fluorescence (TIRF), the researchers reported that the M146L mutation in PS1 prevented STIM1 oligomerization and bungled its hook-up with ORAI-1, thus preventing calcium influx. Again, this effect was abolished when the researchers treated the cells with a γ-secretase inhibitor.
How might mutated PS1 thwart STIM1 oligomerization? The researchers hypothesized that STIM1 was a γ-secretase substrate. Like APP, STIM1 is a type I transmembrane protein, and the amino acid sequence of its transmembrane region is similar to that of APP. In support of their hypothesis, the researchers found a very faint band, representing a potential STIM1 cleavage product, in western blots prepared from cells expressing wild-type or mutant PS1. In an in vitro γ-secretase assay, the M146L-PS1 cleaved STIM1 more efficiently than did the wild-type enzyme. This mutant also cleaved the transmembrane region of APP more efficiently than did wild-type PS1.
To determine whether PS1’s cleavage of STIM1 would exact a physiological toll, the researchers next measured the stability of mature, mushroom-shaped dendritic spines on the surface of hippocampal neurons. The influx of calcium triggered by STIM1 is necessary for the maintenance of these spines, which are crucial for memory. The researchers transfected primary rat hippocampal neurons with wild-type or M146L-PS1, and found that both the density of mature spines and the number of neurites decorated with those spines was lower in cells expressing the mutated version of PS1. When the researchers either overexpressed STIM1 or inhibited γ-secretase, dendritic spine numbers were restored to normal. These results indicated that enhanced cleavage of STIM1 by mutated PS1 reduced calcium influx, leading to the collapse of mature dendritic spines.
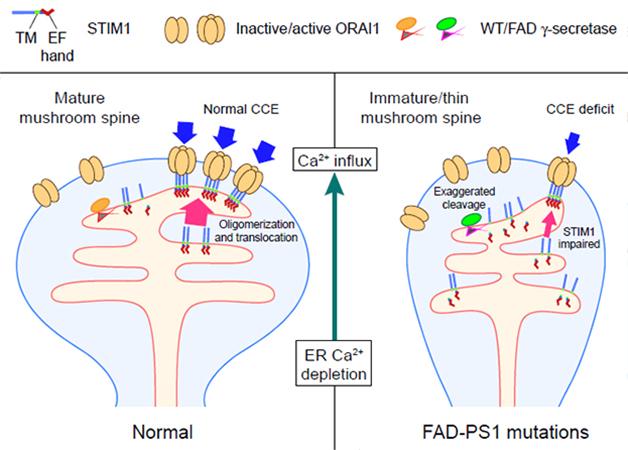
Incapacitated. In cells with normal PS1, STIM1 oligomerization triggers CCE and maintains mature spines. In mutant PS1 neurons, subpar calcium replenishment caused by cleavage of STIM1 destabilizes spines. [Image courtesy of Tong et al., Science Signaling, 2016.]
The findings strengthen the “calcium hypothesis of AD,” first proposed more than two decades ago, commented Jacek Kuznicki of the International Institute of Molecular and Cell Biology in Warsaw (see Khachaturian, 1994). He drew connections with the current paper and the previous observations from Bezprozvanny’s lab that FAD mutations in PS1 decrease expression of STIM2, albeit in a γ-secretase independent fashion. “The common pathophysiological outcome is the reduced number of mushroom spines in FAD PS1 neurons,” he wrote. He added that targeting such calcium entry pathways, rather than Aβ, may be an attractive therapeutic strategy.
Bezprozvanny commented that while the results jibe with those from his lab in some ways, the researchers will need to account for the variable effects of different presenilin mutations. “Many PS1-FAD mutants reduce γ-secretase activity, and it remains to be determined how the proposed model works for these mutants, as they would be expected to have reduced levels of STIM cleavage when compared to the wild type,” he wrote to Alzforum (see full comment below).
Cheung agreed it is unclear how FAD-linked mutations in PS1 would enhance STIM1 cleavage, but speculated that the unique environment of the ER might influence the substrate specificity and activity of the enzyme. He plans to test more PS1 mutations for their effects on STIM1 processing in the future, and also to test the hypothesis that PS1 cleaves STIM2 as well. He added that it was possible the PS1/STIM1 pathway was only relevant in people with certain mutations, although problems with calcium homeostasis are a common feature of both familial and sporadic forms of the disease. Just as researchers are busy designing γ-secretase modulators that preferentially block the enzyme’s cleavage of APP, Cheung proposed a similar strategy to block γ-secretase destruction of STIM1.
Thayer added that alterations in the STIM1 pathway could explain some of the adverse effects of more general γ-secretase inhibitors. “Clearly, effects on STIM will be an important consideration for drug development targeted to the γ-secretase complex,” he wrote.
Mark Mattson of the National Institute on Aging in Bethesda, Maryland, noted, as did the authors, that calcium homeostasis requires a delicate balance. Before laying out therapeutic strategies, researchers will need to further investigate the normal physiological role of PS1 in limiting calcium entry, he suggested. He added that studying the pathway in the context of age-related changes, such as oxidative stress, mitochondrial dysfunction, and neuronal hyperactivity, will be crucial. Creating an AD model mouse with a version of STIM1 that cannot be cleaved by γ-secretase would allow researchers to test the role of the STIM1/PS1 pathway in AD pathogenesis, he said.—Jessica Shugart
References
News Citations
- Presenilins Open Escape Hatch for ER Calcium
- Perplexing Presenilins: New Evidence for Calcium Leak Channels
- Channel Surfing—Two Studies Strengthen Calcium-AD Connection
- Inositol Triphosphate Receptor Blamed for Calcium Chaos in Alzheimer’s
- Calcium Sensor STIM2 Maintains Synapses, Ebbs in Alzheimer’s
Mutations Citations
Paper Citations
- Leissring MA, Akbari Y, Fanger CM, Cahalan MD, Mattson MP, Laferla FM. Capacitative calcium entry deficits and elevated luminal calcium content in mutant presenilin-1 knockin mice. J Cell Biol. 2000 May 15;149(4):793-8. PubMed.
- Yoo AS, Cheng I, Chung S, Grenfell TZ, Lee H, Pack-Chung E, Handler M, Shen J, Xia W, Tesco G, Saunders AJ, Ding K, Frosch MP, Tanzi RE, Kim TW. Presenilin-mediated modulation of capacitative calcium entry. Neuron. 2000 Sep;27(3):561-72. PubMed.
- Cheung KH, Shineman D, Müller M, Cárdenas C, Mei L, Yang J, Tomita T, Iwatsubo T, Lee VM, Foskett JK. Mechanism of Ca2+ disruption in Alzheimer's disease by presenilin regulation of InsP3 receptor channel gating. Neuron. 2008 Jun 26;58(6):871-83. PubMed.
- Cheung KH, Mei L, Mak DO, Hayashi I, Iwatsubo T, Kang DE, Foskett JK. Gain-of-function enhancement of IP3 receptor modal gating by familial Alzheimer's disease-linked presenilin mutants in human cells and mouse neurons. Sci Signal. 2010;3(114):ra22. PubMed.
- Akbari Y, Hitt BD, Murphy MP, Dagher NN, Tseng BP, Green KN, Golde TE, Laferla FM. Presenilin regulates capacitative calcium entry dependently and independently of gamma-secretase activity. Biochem Biophys Res Commun. 2004 Oct 1;322(4):1145-52. PubMed.
- Bojarski L, Pomorski P, Szybinska A, Drab M, Skibinska-Kijek A, Gruszczynska-Biegala J, Kuznicki J. Presenilin-dependent expression of STIM proteins and dysregulation of capacitative Ca2+ entry in familial Alzheimer's disease. Biochim Biophys Acta. 2009 Jun;1793(6):1050-7. Epub 2008 Dec 6 PubMed.
- Khachaturian ZS. Calcium hypothesis of Alzheimer's disease and brain aging. Ann N Y Acad Sci. 1994 Dec 15;747:1-11. PubMed.
Other Citations
Further Reading
Primary Papers
- Tong BC, Lee CS, Cheng WH, Lai KO, Foskett JK, Cheung KH. Familial Alzheimer's disease-associated presenilin 1 mutants promote γ-secretase cleavage of STIM1 to impair store-operated Ca2+ entry. Sci Signal. 2016 Sep 6;9(444):ra89. PubMed.
Annotate
To make an annotation you must Login or Register.
Comments
UT Southwestern Medical Center at Dallas
This is an interesting paper that suggests a potential mechanism for reduction of store-operated calcium entry (SOC) in AD neurons. Our laboratory previously suggested that reduction in SOC calcium entry is one of the potential reasons for synaptic instability in AD neurons. In our study, we observed reduced levels of STIM2 in PS1-M146V KI neurons (Sun et al., 2014). The hypothesis in this paper by Tong at al. is that PS1-M146L and other PS1-FAD mutants are “gain of γ-secretase function” mutants that excessively cut STIM proteins, resulting in defective SOC. Similar to our findings, Tong at al. observed loss of mushroom spines in hippocampal neurons transfected with PS1-M146L mutant, which could be rescued by expression of STIM1 protein. Proposed mechanism may potentially be responsible for reduced levels of STIM proteins and SOC defects in PS1-FAD neurons. However, many PS1-FAD mutants reduce γ-secretase activity (Xia et al., 2015; Bentahir et al., 2006; Fernandez et al., 2014), and it remains to be determined how the proposed model works for these FAD mutants, as they would be expected to have reduced levels of STIM cleavage when compared to the wild type.
References:
Sun S, Zhang H, Liu J, Popugaeva E, Xu NJ, Feske S, White CL 3rd, Bezprozvanny I. Reduced synaptic STIM2 expression and impaired store-operated calcium entry cause destabilization of mature spines in mutant presenilin mice. Neuron. 2014 Apr 2;82(1):79-93. PubMed.
Xia D, Watanabe H, Wu B, Lee SH, Li Y, Tsvetkov E, Bolshakov VY, Shen J, Kelleher RJ 3rd. Presenilin-1 knockin mice reveal loss-of-function mechanism for familial Alzheimer's disease. Neuron. 2015 Mar 4;85(5):967-81. PubMed.
Bentahir M, Nyabi O, Verhamme J, Tolia A, Horré K, Wiltfang J, Esselmann H, De Strooper B. Presenilin clinical mutations can affect gamma-secretase activity by different mechanisms. J Neurochem. 2006 Feb;96(3):732-42. PubMed.
Fernandez MA, Klutkowski JA, Freret T, Wolfe MS. Alzheimer presenilin-1 mutations dramatically reduce trimming of long amyloid β-peptides (Aβ) by γ-secretase to increase 42-to-40-residue Aβ. J Biol Chem. 2014 Nov 7;289(45):31043-52. Epub 2014 Sep 19 PubMed.
International Institute of Molecular and Cell Biology in Warsaw (IIMCB)
The paper brings an interesting mechanistic insight into the action of FAD PS1 on store-operated calcium entry (SOCE). It has long been known that FAD mutations in PS1 lead to a reduction in SOCE, whereas expression of catalytically-inactive PS1 mutants had a converse effect, thereby linking γ-secretase activity with SOCE. Although γ-secretase processes another FAD-linked protein, namely amyloid-precursor protein (APP), the involvement of APP in SOCE is controversial. Tong et al. now report that STIM1, a Ca2+ sensor in the ER, interacts with PS1 and is also processed by γ-secretase. The cleavage is increased in the presence of FAD PS1, leading to a smaller pool of STIM1 molecules that are capable of activating Orai1 plasma membrane calcium channels, explaining the observed effects on SOCE. Strikingly, the overall steady-state levels of STIM1 were hardly changed, which somewhat contrasts the findings of others showing more pronounced effects of PS1 on STIM1 or STIM2 levels (Bojarski et al., 2009; Sun et al., 2014). Despite these differences, a common paradigm appears to be that FAD PS1 attenuates SOCE by affecting STIM Ca2+ sensors, either through γ-secretase-dependent (this paper) or secretase–independent (Sun et al.) mechanisms. The common pathophysiological outcome is the reduced number of mushroom spines in FAD PS1 neurons (Tong et al., Sun et al.).
One can ask how relevant the data are for understanding the mechanisms of calcium disruption in Alzheimer’s disease. In my opinion the data may explain the pathology in familial cases of AD related to mutations in presenilins, but there is no clear mechanism to explain the effect of FAD APP mutations. We (Wegierski et al., 2016) and others found no effect of APP on SOCE. The most interesting outcome of the Tong et al. paper is that it indirectly strengthens the calcium hypothesis of sporadic AD proposed by Khachaturian (1994). The observation by Sun et al. that STIM2 is also decreased in the brains of SAD patients, together with our data showing dysregulation of calcium homeostasis, including SOCE, in fresh human lymphocytes isolated from people with SAD and MCI (Jaworska et al., 2013), indicate that targeting proteins of SOCE instead of Aβ might be an attractive alternative to anti-AD treatment. This is what we suggested in recent reviews (Wojda and Kuznicki, 2013; Majewski and Kuznicki, 2015).
References:
Bojarski L, Pomorski P, Szybinska A, Drab M, Skibinska-Kijek A, Gruszczynska-Biegala J, Kuznicki J. Presenilin-dependent expression of STIM proteins and dysregulation of capacitative Ca2+ entry in familial Alzheimer's disease. Biochim Biophys Acta. 2009 Jun;1793(6):1050-7. Epub 2008 Dec 6 PubMed.
Sun S, Zhang H, Liu J, Popugaeva E, Xu NJ, Feske S, White CL 3rd, Bezprozvanny I. Reduced synaptic STIM2 expression and impaired store-operated calcium entry cause destabilization of mature spines in mutant presenilin mice. Neuron. 2014 Apr 2;82(1):79-93. PubMed.
Wegierski T, Gazda K, Kuznicki J. Microscopic analysis of Orai-mediated store-operated calcium entry in cells with experimentally altered levels of amyloid precursor protein. Biochem Biophys Res Commun. 2016 Sep 23;478(3):1087-92. Epub 2016 Aug 12 PubMed.
Khachaturian ZS. Calcium hypothesis of Alzheimer's disease and brain aging. Ann N Y Acad Sci. 1994 Dec 15;747:1-11. PubMed.
Jaworska A, Dzbek J, Styczynska M, Kuznicki J. Analysis of calcium homeostasis in fresh lymphocytes from patients with sporadic Alzheimer's disease or mild cognitive impairment. Biochim Biophys Acta. 2013 Jan 24; PubMed.
Wojda U, Kuznicki J. Alzheimer's disease modeling: ups, downs, and perspectives for human induced pluripotent stem cells. J Alzheimers Dis. 2013 Jan 1;34(3):563-88. PubMed.
Majewski L, Kuznicki J. SOCE in neurons: Signaling or just refilling?. Biochim Biophys Acta. 2015 Sep;1853(9):1940-52. Epub 2015 Jan 31 PubMed.
Make a Comment
To make a comment you must login or register.