Mislocalized Mitochondria Sufficient for Motor Neuron Malaise
Quick Links
Messing with mitochondrial motility, even if the organelles functioned perfectly, was sufficient to cause neurological disease in mice, according to a paper in the August 18 Proceedings of the National Academy of Sciences online. The work, from the laboratory of Janet Shaw at the University of Utah School of Medicine in Salt Lake City, neatly separates movement of mitochondria from two of their main functions—respiration and calcium buffering. Mice lacking a crucial mitochondrial transport factor are healthy at birth, but rapidly develop an apparent upper motor neuron disorder, and die within a month. The animals develop symptoms reminiscent of amyotrophic lateral sclerosis, hereditary spastic paraplegia, and dystonia.
Novel Mouse Model
Mitochondria rely on kinesin and dynein motors to get around, particularly along motor neurons’ lengthy axons. Mitochondrial Rho (Miro) GTPases are among several cellular factors that attach the organelle to these motors, and act as a sort of brake to arrest the cellular powerhouse where they are most needed, for example close to synapses. To examine the relationship between mitochondrial motions and mammalian disease, first author Tammy Nguyen engineered mice with a floxed Miro1 sequence, which she could then delete by introducing the enzyme Cre. First, she made mice that lost Miro1 during the embryo stage. The animals were fine in utero, but upon birth failed to take a single breath.
The authors investigated the nerve circuits required for that first gasp and found the knockouts were missing crucial elements, including motor neurons in a cranial structure called the nucleus ambiguus. They also had a dearth of the spinal cord motor neurons that control the diaphragm muscle, and the phrenic nerve, which runs from the cord to the diaphragm, had fewer branches than it did in wild-type mice (see image below). The authors surmised that their Miro1 knockouts died because of motor neuron defects.
In Miro1 knockout mice (bottom), the phrenic nerve that controls diaphragm muscle contraction lacked the normal branches found in wild-type animals (top). [Image courtesy of Nguyen et al., PNAS.]
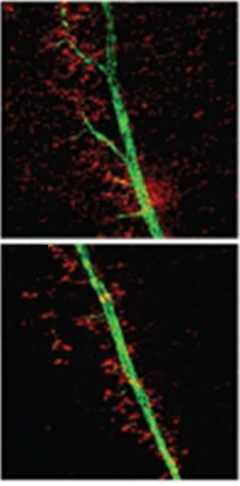
The authors noted these symptoms resembled upper motor neuron disease, and examined the animals’ spinal cords for markers of neuropathology. They noticed aggregates in the knockout mouse tissues that were reminiscent of Bunina bodies, poorly characterized inclusions commonly observed in the spinal cords of people who died of ALS. However, Bunina bodies are found inside cells, while the aggregates in the Miro1 knockout mice appeared to be extracellular. They might be the remains of dead neurons, the authors suggest.
Do the mice model any particular human disease? At this point, the researchers are uncertain. Because their problems started so early, Shaw initially thought the mice mimicked hereditary spastic paraplegia, a developmental condition. However, she thinks she may be able to extend the animals’ lifespan by placing their food within easier reach, in which case they might model ALS. Some of her colleagues have suggested that the symptoms more closely resemble dystonia. “There is still a lot of characterization to do,” Shaw said. “We need to know more about what is going on in the mouse so we can better relate it to the human condition.”
Serge Przedborski of Columbia University in New York, who was not involved in the study, said it was difficult to determine whether the mice correspond to any human disease. For one, he noted, the animals are only missing Miro1 in certain neurons. He prefers germline models in which all cells share the same genetic defect, such as the mutant superoxide dismutase 1 mice that mimic human ALS. In these mice as well as in people who have the same disease gene, all cells carry the mutation but only motor neurons degenerate.
Przedborski added that spasticity in rodents results from different neurological pathways than it does in people, so the symptoms could be species-specific. Similarly, he said, it is difficult to interpret the Bunina-like bodies because they are not intracellular as in people with ALS.
To understand Miro1’s role better, the authors are planning a new mouse model with Cre controlled by an inducible promoter. With that system, they can remove Miro1 selectively in older animals and potentially create a degenerative condition like ALS. In addition, they are screening people with inherited neurological diseases of unknown genetic origin, looking for Miro1 mutations.
Mitochondrial Stumbles
Regardless of whether they mimic any specific human disease, the mice allowed Nguyen and Shaw to probe how mitochondrial mobility relates to neuron health. Numerous studies have linked defective mitochondria to neurodegeneration (see Jun 2014 news story, news story), but the mitochondria usually exhibit problems both in respiration and localization. However, loss of Miro1 apparently affected only transport, not function.
The mitochondria from the Miro1-negative neurons behaved normally in several assays. In primary cortical neuron cultures, the mitochondrial membrane potential was normal. Nguyen also isolated embryonic fibroblasts from the original knockout mice, lacking Miro1 in every cell, and measured rates of oxygen consumption. Oxygen uptake was also typical in that mitochondria do not need Miro1 for respiration.
Mitochondria also prevent calcium toxicity by buffering the cation, and Miro1 contains several calcium-binding motifs. However, the Miro1-negative fibroblasts managed calcium levels properly.
The only defect Nguyen found was in mitochondrial motion. Though the organelles lacking Miro 1 were able to travel, they exhibited specific defects. In the spinal cord axons of the knockout mice, Nguyen observed fewer than normal mitochondria, indicating improper distribution of the organelles. Using a microscope, she observed that the mitochondria of Miro1-negative cells stalled more often. Specifically, their overall retrograde speed was one-fourth of normal, though their anterograde motion was fine.
“This is the first demonstration that you can cause neurological disease simply by preventing mitochondria from moving to the right place, even if they are functioning,” Shaw concluded. She suggested this model might be useful to test drugs that redistribute the organelle properly. Such medications might see widespread use, Shaw speculated, since many neurological conditions, including Alzheimer’s, appear to involve defective mitochondrial distribution (see Feb 2010 news story, Jul 2009 news story, Apr 2009 news story).—Amber Dance.
References
News Citations
- Mitochondrial Mutation Linked to Syndrome With ALS-FTD Features
- No MAM: ALS Protein Breaks Mitochondria-Endoplasmic Reticulum Bond
- Abnormal Mitochondrial Dynamics—Early Event in AD, PD?
- Mitochondrial Break-up: Alzheimer’s Alters Fusion, Fission
- NO Kidding? Mitochondria Fission Protein Linked to Neurodegeneration
Further Reading
Papers
- Kopeikina KJ, Carlson GA, Pitstick R, Ludvigson AE, Peters A, Luebke JI, Koffie RM, Frosch MP, Hyman BT, Spires-Jones TL. Tau accumulation causes mitochondrial distribution deficits in neurons in a mouse model of tauopathy and in human Alzheimer's disease brain. Am J Pathol. 2011 Oct;179(4):2071-82. PubMed.
- Wang X, Su B, Fujioka H, Zhu X. Dynamin-like protein 1 reduction underlies mitochondrial morphology and distribution abnormalities in fibroblasts from sporadic Alzheimer's disease patients. Am J Pathol. 2008 Aug;173(2):470-82. PubMed.
- Sotelo-Silveira JR, Lepanto P, Elizondo V, Horjales S, Palacios F, Martinez-Palma L, Marin M, Beckman JS, Barbeito L. Axonal mitochondrial clusters containing mutant SOD1 in transgenic models of ALS. Antioxid Redox Signal. 2009 Jul;11(7):1535-45. PubMed.
- Esteves AR, Gozes I, Cardoso SM. The rescue of microtubule-dependent traffic recovers mitochondrial function in Parkinson's disease. Biochim Biophys Acta. 2014 Jan;1842(1):7-21. Epub 2013 Oct 11 PubMed.
- Gan X, Huang S, Wu L, Wang Y, Hu G, Li G, Zhang H, Yu H, Swerdlow RH, Chen JX, Yan SS. Inhibition of ERK-DLP1 signaling and mitochondrial division alleviates mitochondrial dysfunction in Alzheimer's disease cybrid cell. Biochim Biophys Acta. 2014 Feb;1842(2):220-31. Epub 2013 Nov 16 PubMed.
- Bonda DJ, Wang X, Perry G, Smith MA, Zhu X. Mitochondrial dynamics in Alzheimer's disease: opportunities for future treatment strategies. Drugs Aging. 2010 Mar 1;27(3):181-92. PubMed.
News
- Studies Suggest Mitochondria Changes Precede Aging, Alzheimer’s
- Energy Crisis: ATP Deficiency Dooms Motor Neurons in Computer Model
- Parkinsonism-linked Protein Binds Parkin and Pink1, Drives Mitophagy
- Protein Destroying Muscle, Bone, Nerves Parks on Mitochondria
- Could Too Much Tau Be a Stretch for Mitochondria?
- Sluggish Mitochondria Not Causing Withered ALS Axons After All?
Primary Papers
- Nguyen TT, Oh SS, Weaver D, Lewandowska A, Maxfield D, Schuler MH, Smith NK, Macfarlane J, Saunders G, Palmer CA, Debattisti V, Koshiba T, Pulst S, Feldman EL, Hajnóczky G, Shaw JM. Loss of Miro1-directed mitochondrial movement results in a novel murine model for neuron disease. Proc Natl Acad Sci U S A. 2014 Aug 18; PubMed.
Annotate
To make an annotation you must Login or Register.
Comments
Case Western Reserve University
It is generally believed that the proper intracellular distribution of mitochondria, adapting to localized bioenergetic requirement, is critical for cellular physiology (Frazier et al., 2006). Despite previous studies nicely showing the regulation of mitochondrial mobility/distribution by Miro-1 in in vitro cultured cells (Saotome et al., 2008; Macaskill et al., 2009; Wang et al., 2009), the functional role of Miro-1 in vivo remains elusive.
This elegant study, for the first time, provides clear and convincing evidence that Miro-1 was specifically required for upper motor neuron development and survival in mice in vivo. A critical question relevant to this study is why upper motor neurons are vulnerable to Miro-1 ablation. It would be interesting to know if the loss of Miro-1 in other types of cells, such as cortical neurons and muscle, is also sufficient to impair mitochondrial distribution/movement and cause cellular dysfunction in vivo.
Mitochondrial movement deficits and altered mitochondrial distribution have been repeatedly reported in experimental models of motor neuron disease such as amyotrophic lateral sclerosis (ALS) (Magrane et al., 2014; Wang et al., 2013; Bilsland et al., 2010; De Vos et al., 2007). In this study, the authors showed that the neuronal specific loss of Miro-1 caused symptoms of motor neuron diseases, strongly supporting the possibility that mitochondrial distribution/movement deficits might be the cause rather than the consequence of motor neuron diseases. Miro-1 is a mitochondrial protein specifically regulating mitochondrial movement, and the in vivo system used in this study is relatively clean, especially considering the authors provided multiple lines of evidence demonstrating unchanged mitochondrial functions by Miro-1 ablation.
Growing evidence suggests a critical role of mitochondria in the pathogenesis of neurodegenerative diseases including ALS, Alzheimer’s disease, and Parkinson’s disease. Although it still remains unclear whether Miro-1 or the machinery regulating mitochondrial movement/distribution is indeed altered in patients and experimental models of neurodegenerative diseases, the Miro-1 floxed animal model used in this study is very useful and could be used to test whether and how the altered mitochondrial distribution contribute to disease onset and progression.
References:
Frazier AE, Kiu C, Stojanovski D, Hoogenraad NJ, Ryan MT. Mitochondrial morphology and distribution in mammalian cells. Biol Chem. 2006 Dec;387(12):1551-8. PubMed.
Saotome M, Safiulina D, Szabadkai G, Das S, Fransson A, Aspenstrom P, Rizzuto R, Hajnóczky G. Bidirectional Ca2+-dependent control of mitochondrial dynamics by the Miro GTPase. Proc Natl Acad Sci U S A. 2008 Dec 30;105(52):20728-33. Epub 2008 Dec 19 PubMed.
Macaskill AF, Rinholm JE, Twelvetrees AE, Arancibia-Carcamo IL, Muir J, Fransson A, Aspenstrom P, Attwell D, Kittler JT. Miro1 is a calcium sensor for glutamate receptor-dependent localization of mitochondria at synapses. Neuron. 2009 Feb 26;61(4):541-55. PubMed.
Wang X, Schwarz TL. The mechanism of Ca2+ -dependent regulation of kinesin-mediated mitochondrial motility. Cell. 2009 Jan 9;136(1):163-74. PubMed.
Magrané J, Cortez C, Gan WB, Manfredi G. Abnormal mitochondrial transport and morphology are common pathological denominators in SOD1 and TDP43 ALS mouse models. Hum Mol Genet. 2014 Mar 15;23(6):1413-24. Epub 2013 Oct 23 PubMed.
Wang W, Li L, Lin WL, Dickson DW, Petrucelli L, Zhang T, Wang X. The ALS disease-associated mutant TDP-43 impairs mitochondrial dynamics and function in motor neurons. Hum Mol Genet. 2013 Dec 1;22(23):4706-19. Epub 2013 Jul 4 PubMed.
Bilsland LG, Sahai E, Kelly G, Golding M, Greensmith L, Schiavo G. Deficits in axonal transport precede ALS symptoms in vivo. Proc Natl Acad Sci U S A. 2010 Nov 23;107(47):20523-8. Epub 2010 Nov 8 PubMed.
De Vos KJ, Chapman AL, Tennant ME, Manser C, Tudor EL, Lau KF, Brownlees J, Ackerley S, Shaw PJ, McLoughlin DM, Shaw CE, Leigh PN, Miller CC, Grierson AJ. Familial amyotrophic lateral sclerosis-linked SOD1 mutants perturb fast axonal transport to reduce axonal mitochondria content. Hum Mol Genet. 2007 Nov 15;16(22):2720-8. Epub 2007 Aug 28 PubMed.
Make a Comment
To make a comment you must login or register.