Lysosomal Diseases: Stepping Stones to Gene Therapy for Alzheimer’s?
Quick Links
Gene therapy has re-emerged as a potentially revolutionary treatment for familial and, eventually, sporadic, Alzheimer’s disease (Parts 2 and 3 of the Society for Neuroscience 2019 annual meeting series). While researchers are mulling over targets and strategies, what can they learn from gene therapies that are already in, or close to, clinical trials? Dozens of such therapies are being developed for lysosomal dysfunction, which is a feature of Alzheimer’s, Parkinson’s, and frontotemporal dementias (Whyte et al., 2017; Bonam et al., 2019). Could treatments for lysosomal storage disorders help scientists find ways to rid aging cells of proteins that lead to neurodegeneration?
- Lysosomal dysfunction is common in neurodegenerative disease.
- Childhood lysosomal storage disorders are poised for gene therapy trials.
- Can scientists learn from them to develop Alzheimer’s therapies?
It’s clear that cells need well-functioning lysosomes to maintain homeostasis. These acidic organelles are brimming with enzymes ready to break down washed-up proteins, lipids, and carbohydrates that end up inside lysosomes when they fuse with autophagosomes. The little sacs are deceptively complex: a typical lysosome contains around 60 different acid hydrolases alone, cathepsins being perhaps the best-known, plus granulins and other components (Marques and Saftig 2019).
Lysosomes degrade Aβ, α-synuclein, and other proteins that accumulate in neurodegenerative disorders (Wang et al., 2012; Shacka et al., 2008). In the AD brain, lysosomes accumulate in neurons, and lysosomal proteins are even found in amyloid plaques (Nixon et al., 1992; Ihara et al., 2012). Since then, a steady clip of papers have documented the importance of lysosomal degradation in age-related neurodegeneration. Some implicated microglia (Paresce et al., 1997; Solé-Domènech et al., 2016). Most recently, single-cell analysis of AD brain tissue pinpointed upregulation of the lysosomal master transcription factor TFEB in astrocytes, where it regulates expression of as many as 10 AD genes (Nov 2019 news). Scientists believe that when lysosomes malfunction in subtle ways, proteins such as Aβ, α-synuclein, and TDP-43 can gradually build up in cells of the aging brain, increasing the chances they will form toxic oligomers or aggregates.
But when lysosomes take a severe hit, disease strikes already in childhood. Scientists know of at least nine forms of lysosomal storage disorder, including Niemann-Pick type C, Gaucher, Tay-Sachs, and Fabry diseases. Many have subtypes. The 14 known neuronal ceroid lipofuscinoses, seven mucopolysaccharidoses, five mucolipidoses, and two gangliosidoses are named after the type of molecule whose defective degradation in lysosomes leads to their buildup, i.e., storage.
Interestingly for gene-therapy developers, almost all these lysosomal storage diseases are monogenetic, caused by a pathogenic mutation in one or two gene copies encoding an enzyme or protein essential to lysosome function. They tend to start in infancy, childhood, or young adulthood and lead to physical deformities, dementia, motor defects, sometimes blindness, and often an early death (Kohlschütter et al., 2019).
Heterozygous or mild loss-of-function mutations in some of the affected genes cause late-onset neurodegeneration. For example, mild mutations in glucocerebrosidase increase risk for PD, whereas complete loss causes Gaucher’s. Mild loss-of-function variants in progranulin—which is processed into smaller granulins crucial for lysosome function—cause FTD, whereas complete loss causes a fatal childhood neuronal ceroid lipofuscinosis (NCL) (Sep 2017 news; Götzl et al., 2014; Arrant et al., 2018).
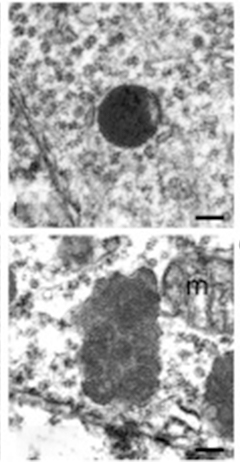
Lysosomal Indigestion. Unlike smooth, round lysosomes in progranulin heterozygote mice (top), lysosomes in progranulin knockouts (bottom) accumulate undigested membrane, making them bumpy and swollen. [Courtesy of Cell Reports, Evers et al. 2017.]
Gene Therapy for Lysosomes
A missing enzyme is an open invitation to gene therapists. In theory, if a vector carrying the gene for that enzyme can be safely delivered to the right place and stably expressed there, then a single injection could stop, even cure, the disease. The spinal muscular atrophy gene therapy zolgensma serves as proof of principle (Part 2 of SfN series). At this point, several academic centers and at least 10 companies are poised to test this concept in dozens of lysosomal storage disorders, including NCLs and mucopolysaccharidoses (MPS). In the former, lysosomes get clogged by lipofuscin, a pigment made up of fat and protein. In the latter, lysosomes can’t break down glycosaminoglycans, which accumulate in neurons and other cells and then impede degradation of other unwanted macromolecules.
Of the seven recognized forms of mucopolysaccharidoses, MPS III, aka Sanfilippo syndrome, is caused by loss-of-function mutations in N-sulfoglucosamine sulfohydrolase (SGSH). Furthest along in clinical testing for this lysosomal storage disorder is LYS-SAF302, developed by Lysogene, Neuilly-sur-Seine, France, in collaboration with Sarepta Therapeutics, Cambridge, Massachusetts. LYS-SAF302 carries normal copies of SGSH on an AAVrh10 vector developed by REGENXBIO, Rockville, Maryland. A Phase 2/3 trial of 20 patients began in December 2018 and is expected to finish in January 2022.
The companies Abeona Therapeutics, headquartered in New York City, Esteve in Barcelona, and Phoenix Nest Inc. in Brooklyn, New York, also have clinical programs targeting Sanfilippo syndrome. Abeona’s Phase 1/2 trial of ABO-102, an AAV9 carrying the SGSH gene, began last October and will run until 2022. Esteve’s EGT-101 AAV9 is also in Phase 1/2 (for details, scroll down on the Spanish registry page). Phoenix Nest has licensed a novel AAV variant, AAV-TT, which was shown to better distribute throughout the brain and more efficiently correct MPSIII-like disease in mice and rats than AAV9-based treatments (Tordo et al., 2018). Amicus Therapeutics, Cranbury, New Jersey, has a therapy for MPSIII in preclinical development.

Lysosome Lipofuscin. Confocal microscopy of a human motor neuron shows accumulation of lipofuscin granules (blue and yellow). [Courtesy of McIlwain et al., 2005.]
Researchers are targeting GM1 gangliosidosis, as well. In this disease, sugars build up in the lysosome due to mutations in the β-galactosidase gene GLB1. Its type I form strikes by 6 months, type II forms by 18 months to five years; type III, after 12 years of age. Infants with type I die as young children, kids with type II and III can live to be adults. Besides damaging the liver, spleen, bones and muscles, GM1 gangliosidosis causes intellectual and movement disabilities. Lysogene has begun site selection for a Phase 1/2 trial of LYS-GM101, according to the company website. Passage Bio in Philadelphia has licensed AAV vectors from the University of Pennsylvania to develop gene therapies for GM1 gangliosidosis and frontotemporal dementia. Similarly, Amicus is collaborating with Nationwide Children’s Hospital, Columbus, Ohio, to develop therapies for Tay-Sachs disease, aka GM2 gangliosidosis, caused by mutations in beta-hexosaminidase A. Tay-Sachs comes in three forms that vary by age of onset and severity, with its attendant neurodegeneration causing seizures, learning disabilities, and weakness.
On to neuronal ceroid lipofuscinoses. Its various forms go by “Batten’s disease,” and gene therapies are in the works. Researchers at Cornell University started a Phase 1 trial in 10 children in 2004. Slated to complete in June 2020, it delivers the CLN2 gene for tripeptidyl peptidase on an AAV2 vector. Preliminary results suggest a slowing of disease (Worgall et al., 2008). These researchers began a second Phase 1 of 25 patients in 2010 and a Phase 1/2 trial of eight patients in 2011, both delivering the same CLN2 gene on an AAVrh10 vector. Led by Ron Crystal of Weill Cornell Medicine in New York, the same group started a trial in symptomatic Alzheimer’s, albeit not targeting the lysosome. They will inject an ApoE2 expression vector into the brain to counteract the effects of ApoE4 (Part 2 of SfN series).
But back to Batten’s. Abeona filed an investigational new drug application for ABO-202 in May 2019 and is planning a Phase 1/2. ABO-202 uses the AAV9 vector to deliver CLN1 to the central nervous system. CLN1 encodes palmitoyl protein thioesterase 1, which is essential for clearing lipids and proteins from the lysosome. Lipids accumulate in microglia in AD (Aug 2019 news). Similarly, ABO-201 is on track for trials; this AAV9-based construct delivers CLN3, the gene encoding the transmembrane protein battenin.
For its part, Amicus is developing AAV treatments for four types of Batten disease: CLN1, 3, 6 and 8. Vectors bearing CLN1 and 8 payloads are in animal studies, while a Phase 1/2 of AT-GX-502 for CLN3 started in November 2018. It is treating seven children between the ages of 3 and 10 with a spinal infusion of a healthy copy of the battenin gene on an AAV9 vector.
For 13 people with CLN6 Batten disease, a Phase 1/2 trial of AT-GTX-501 started in 2016. Participants are receiving a single dose of AAV9 carrying CLN6, which encodes an endoplasmic reticulum protein. According to preliminary results of eight patients presented at the 48th annual Child Neurology Society meeting in October 2019, they tolerated AT-GTX-501 well and seemed to progress more slowly, or not at all, compared with historical data from untreated siblings. Up to two years after the infusion, four participants were stable or had improved, three had worsened somewhat. The younger a participant was at the time of gene therapy, the more he or she seemed to benefit, with progression apparently halted in treated infants but merely slowed in 6-year-olds. The trial is expected to run through 2021.
The dementing tauopathy Nieman-Pick’s type C is caused by mutations in NPC1 or NPC2, whose proteins flush cholesterol from lysosomes (Love et al., 1995; Wheeler and Sillence, 2019). Amicus has a preclinical gene therapy for this disease. In France, researchers ran a Phase 1/2 for metachromatic leukodystrophy, which depletes myelin. Their therapy targets buildup of sulfatides in lysosomes by delivering the arylsulfatase A gene on an AAVrh10 vector.
Also under study are gene therapies for lysosomal storage diseases that harm the brain less grievously. Amicus and UPenn are collaborating to treat Fabry and Pompe. In the latter, loss of acid alpha-glucosidase causes glycogen to accumulate in lysosomes throughout the body, but the heart and skeletal muscles take a bigger hit than the brain. The culprit in Fabry is alpha-galactosidase, without which lysosomes get packed with sphingolipids. This disease damages the kidney, heart, and skin, and causes peripheral neuropathic pain.
None of these therapies target AD or related disorders. Still, scientists believe they will learn about lysosomes from these studies, and perhaps figure out ways to boost the function of these organelles in complex age-related proteinopathies.
Last but not least, gene therapies are being deployed against non-lysosomal neurologic disorders, for example to strengthen muscles in Duchenne muscular dystrophy. A 12-patient Phase 1 of an AAV9 carrying a mini dystrophin gene began July 2018, for a six-year run time. A 40-patient Phase 2 of a mini dystrophin vector began in December 2018. Sarepta and Nationwide Children’s Hospital have an AAV carrying GALGT2 in Phase 1/2. This gene encodes β 1,4 –N-acetylgalactosamine galactosyltransferase, which post-translationally modifies proteins at the neuromuscular junction. Sarepta has six other gene therapies in the pipeline for muscular dystrophies that affect the limb muscles closest to the body.
For the myelin disease Charcot-Marie-Tooth, Sarepta will test a vector carrying neurotrophin 3, and, working with REGENXBIO, Pfizer has a preclinical program for Friedreich’s ataxia. MeiraGTx has seen some positive data from its Phase 1/2 trial of AAV-RPE65, which targets retinal dystrophy. And the list goes on.—Tom Fagan and Gabrielle Strobel
References
News Citations
- Time to Try Again: Gene-Based Therapy for Neurodegeneration
- Gene Therapies Enter Trials for Many Brain Pathologies—What about AD?
- Single-Cell Expression Atlas Charts Changes in Alzheimer’s Entorhinal Cortex
- Lysosomes Take Center Stage in Parkinson’s and Frontotemporal Dementia
- Newly Identified Microglia Contain Lipid Droplets, Harm Brain
Paper Citations
- Whyte LS, Lau AA, Hemsley KM, Hopwood JJ, Sargeant TJ. Endo-lysosomal and autophagic dysfunction: a driving factor in Alzheimer's disease?. J Neurochem. 2017 Mar;140(5):703-717. Epub 2017 Jan 23 PubMed.
- Bonam SR, Wang F, Muller S. Lysosomes as a therapeutic target. Nat Rev Drug Discov. 2019 Dec;18(12):923-948. Epub 2019 Sep 2 PubMed.
- Marques AR, Saftig P. Lysosomal storage disorders - challenges, concepts and avenues for therapy: beyond rare diseases. J Cell Sci. 2019 Jan 16;132(2) PubMed.
- Wang C, Sun B, Zhou Y, Grubb A, Gan L. Cathepsin B Degrades Amyloid-β in Mice Expressing Wild-type Human Amyloid Precursor Protein. J Biol Chem. 2012 Nov 16;287(47):39834-41. PubMed.
- Shacka JJ, Roth KA, Zhang J. The autophagy-lysosomal degradation pathway: role in neurodegenerative disease and therapy. Front Biosci. 2008;13:718-36. PubMed.
- Nixon RA, Cataldo AM, Paskevich PA, Hamilton DJ, Wheelock TR, Kanaley-Andrews L. The lysosomal system in neurons. Involvement at multiple stages of Alzheimer's disease pathogenesis. Ann N Y Acad Sci. 1992 Dec 31;674:65-88. PubMed.
- Ihara Y, Morishima-Kawashima M, Nixon R. The ubiquitin-proteasome system and the autophagic-lysosomal system in Alzheimer disease. Cold Spring Harb Perspect Med. 2012;2(8) PubMed.
- Paresce DM, Chung H, Maxfield FR. Slow degradation of aggregates of the Alzheimer's disease amyloid beta-protein by microglial cells. J Biol Chem. 1997 Nov 14;272(46):29390-7. PubMed.
- Solé-Domènech S, Cruz DL, Capetillo-Zarate E, Maxfield FR. The endocytic pathway in microglia during health, aging and Alzheimer's disease. Ageing Res Rev. 2016 Dec;32:89-103. Epub 2016 Jul 12 PubMed.
- Kohlschütter A, Schulz A, Bartsch U, Storch S. Current and Emerging Treatment Strategies for Neuronal Ceroid Lipofuscinoses. CNS Drugs. 2019 Apr;33(4):315-325. PubMed.
- Götzl JK, Mori K, Damme M, Fellerer K, Tahirovic S, Kleinberger G, Janssens J, van der Zee J, Lang CM, Kremmer E, Martin JJ, Engelborghs S, Kretzschmar HA, Arzberger T, Van Broeckhoven C, Haass C, Capell A. Common pathobiochemical hallmarks of progranulin-associated frontotemporal lobar degeneration and neuronal ceroid lipofuscinosis. Acta Neuropathol. 2014 Mar 12; PubMed.
- Arrant AE, Onyilo VC, Unger DE, Roberson ED. Progranulin Gene Therapy Improves Lysosomal Dysfunction and Microglial Pathology Associated with Frontotemporal Dementia and Neuronal Ceroid Lipofuscinosis. J Neurosci. 2018 Feb 28;38(9):2341-2358. Epub 2018 Jan 29 PubMed.
- Tordo J, O'Leary C, Antunes AS, Palomar N, Aldrin-Kirk P, Basche M, Bennett A, D'Souza Z, Gleitz H, Godwin A, Holley RJ, Parker H, Liao AY, Rouse P, Youshani AS, Dridi L, Martins C, Levade T, Stacey KB, Davis DM, Dyer A, Clément N, Björklund T, Ali RR, Agbandje-McKenna M, Rahim AA, Pshezhetsky A, Waddington SN, Linden RM, Bigger BW, Henckaerts E. A novel adeno-associated virus capsid with enhanced neurotropism corrects a lysosomal transmembrane enzyme deficiency. Brain. 2018 Jul 1;141(7):2014-2031. PubMed.
- Worgall S, Sondhi D, Hackett NR, Kosofsky B, Kekatpure MV, Neyzi N, Dyke JP, Ballon D, Heier L, Greenwald BM, Christos P, Mazumdar M, Souweidane MM, Kaplitt MG, Crystal RG. Treatment of late infantile neuronal ceroid lipofuscinosis by CNS administration of a serotype 2 adeno-associated virus expressing CLN2 cDNA. Hum Gene Ther. 2008 May;19(5):463-74. PubMed.
- Love S, Bridges LR, Case CP. Neurofibrillary tangles in Niemann-Pick disease type C. Brain. 1995 Feb;118 ( Pt 1):119-29. PubMed.
- Wheeler S, Sillence DJ. Niemann-Pick type C disease: cellular pathology and pharmacotherapy. J Neurochem. 2019 Oct 14; PubMed.
External Citations
Further Reading
Papers
- Fraldi A, Serafini M, Sorrentino NC, Gentner B, Aiuti A, Bernardo ME. Gene therapy for mucopolysaccharidoses: in vivo and ex vivo approaches. Ital J Pediatr. 2018 Nov 16;44(Suppl 2):130. PubMed.
- Jiang P, Gan M, Yen SH, McLean PJ, Dickson DW. Impaired endo-lysosomal membrane integrity accelerates the seeding progression of α-synuclein aggregates. Sci Rep. 2017 Aug 9;7(1):7690. PubMed.
Annotate
To make an annotation you must Login or Register.
Comments
Rowan University, NJ Institute for Successful Aging
Ever since Anne Cataldo and Ralph Nixon and their many coworkers (one article was cited above) advanced this line of research over 30 years ago, it has been very clear that lysosomal trafficking is critical to the pathology of Alzheimer's disease. The real question is why this observation has not led to therapies, and why gene therapy has lagged decades behind basic science discoveries.
I believe that the reason is twofold: (1) NIH, which funds most biomedical research in the U.S., has not made human gene therapy a proper priority. (2) Many investors grew "cold feet" for gene therapy in the late 1990s, and only in the last 10 years or so has biopharma interest in viral vectors and antisense therapies steadily risen.
The technology is now mature, and we are in the middle of a renaissance in human gene therapy. Few people were paying attention in the Alzheimer's field, but human gene therapy has re-emerged as a viable approach for breakthrough drugs. A number of companies have been advancing clinical-stage gene therapies for rare monogenic diseases, some of which involve lysosomal storage failure.
The parallels with Alzheimer's disease are intriguing, especially for tauopathies such as NPC. It will be interesting to see how these studies for rare diseases will translate into new avenues for Alzheimer's gene therapy in years to come.
New York University School of Medicine/Nathan Kline Institute
The expanded research on LSDs is exciting and has great potential to inform therapeutic approaches for Alzheimer’s disease (AD) and aging-related neurodegenerative diseases. Urgency and accumulated evidence also justify accelerated efforts now toward targeting the lysosomal system (endocytic and autophagy pathways) in AD.
Evidence, only briefly cited here, shows that endosomal-lysosomal (E-L) dysfunction arises very early in all forms of AD. E-L dysfunction is accelerated by major AD genes (PSEN1/2, APP, APOE4), is progressive throughout the disease course, and is uniquely robust in AD relative to other aging-related neurodegenerative diseases (Nixon, 2017; Nixon and Cataldo, 2006).
Defective lysosomal proteolysis in AD underlies the autophagy failure that leads to hallmark neuritic dystrophy, Aβ accumulation, and a lysosomal-associated form of neuronal cell death in AD (Bordi et al., 2016; Nixon, 2013). Selectively restoring lysosomal function using any of a diversity of genetic and pharmacological means ameliorates amyloidogenesis, synaptic failure, and cognitive decline in AD models (Nixon, 2013; Sharma et al., 2018; Boland et al., 2018).
Why the AD field has been slow to exploit this direction, as Christopher Janson asked in his commentary, has several possible explanations. Not the least among them are lingering misconceptions that major AD genes (PSEN1 and 2, APP, APOE4) implicate only Aβ in pathogenesis and have no direct association with the lysosomal system; thus, all E-L pathobiology in AD is viewed as downstream of Aβ neurotoxicity.
These positions are obsolete from many empirical perspectives. The proteins encoded by these genes, and many GWAS AD risk genes, in fact, are all constituents of the E-L system and modulate its physiological functions. Moreover, in AD, mutant or metabolic forms of APP, PSEN1/2, and ApoE4 interact directly with specific E-L pathway components and disrupt global E-L functions, both independently of Aβ and by impeding lysosomal clearance of lysosomal substrates, including the multiple neurotoxic metabolites of APP (Nixon, 2017; Boland et al., 2018; Lee et al., 2010; Sharma et al., 2019; Im et al., 2019; Jiang et al., 2019).
We have proposed that endosomal-lysosomal dysfunction and the pathogenicity exerted by multiple APP metabolites are inextricably connected genetically and functionally in AD. Through this partnership, they provide a more comprehensive pathobiological explanation for AD and especially for the sporadic forms (Nixon, 2017).
Previously challenged notions, such as direct pathogenic actions of APP-βCTF and neurotoxicity of intralysosomal Aβ/ APP-βCTF arising from E-L dysfunction, now can hardly be considered controversial given the accumulated evidence. The essential direct role of Presenilin 1 in acidifying lysosomes by supporting vATPase assembly, and disruption of this process by PSEN1 mutations or deletion (Lee et al., 2010) sparked a bit of controversy initially, but these findings have now been repeatedly validated (e.g., Lee et al., 2015; Wolfe et al., 2013; Sharma et al., 2019; Martin-Maestro et al., 2017). Indeed, primary vATPase dysfunction as the cause of lysosomal system failure in neurodegenerative disease is a rapidly emerging theme (Colacurcio and Nixon, 2016; Jiang et al., 2019; Im et al., 2019; Wallings et al., 2019).
Overlaps exist also with certain LSDs. Niemann-Pick type C disease shares a remarkable number of pathological features with AD (Cabeza et al., 2012). A pathogenic E-L mechanism analogous to that for PSEN1 in FAD is reported for the CLN1 protein causing neuronal ceroid lipofuscinosis (NCL) (Bagh et al., 2017).
Finally, the extensive pleiotropy of PSEN1 mutations causing other neurodegenerative diseases linked to lysosomal dysfunction (Larner et al., 2013), including Parkinson’s disease, spastic paraparesis, and even an early onset dementia with pathological features of the adult-onset NCL Kufs disease, is more easily understood from the perspective of lysosomal dysfunction than from β-amyloidosis.
References:
Nixon RA. Amyloid precursor protein and endosomal-lysosomal dysfunction in Alzheimer's disease: inseparable partners in a multifactorial disease. FASEB J. 2017 Jul;31(7):2729-2743. PubMed.
Nixon RA, Cataldo AM. Lysosomal system pathways: genes to neurodegeneration in Alzheimer's disease. J Alzheimers Dis. 2006;9(3 Suppl):277-89. PubMed.
Bordi M, Berg MJ, Mohan PS, Peterhoff CM, Alldred MJ, Che S, Ginsberg SD, Nixon RA. Autophagy flux in CA1 neurons of Alzheimer hippocampus: Increased induction overburdens failing lysosomes to propel neuritic dystrophy. Autophagy. 2016 Dec;12(12):2467-2483. Epub 2016 Nov 4 PubMed.
Nixon RA. The role of autophagy in neurodegenerative disease. Nat Med. 2013 Aug;19(8):983-97. PubMed.
Sharma J, di Ronza A, Lotfi P, Sardiello M. Lysosomes and Brain Health. Annu Rev Neurosci. 2018 Jul 8;41:255-276. Epub 2018 Apr 16 PubMed.
Boland B, Yu WH, Corti O, Mollereau B, Henriques A, Bezard E, Pastores GM, Rubinsztein DC, Nixon RA, Duchen MR, Mallucci GR, Kroemer G, Levine B, Eskelinen EL, Mochel F, Spedding M, Louis C, Martin OR, Millan MJ. Promoting the clearance of neurotoxic proteins in neurodegenerative disorders of ageing. Nat Rev Drug Discov. 2018 Sep;17(9):660-688. Epub 2018 Aug 17 PubMed.
Lee JH, Yu WH, Kumar A, Lee S, Mohan PS, Peterhoff CM, Wolfe DM, Martinez-Vicente M, Massey AC, Sovak G, Uchiyama Y, Westaway D, Cuervo AM, Nixon RA. Lysosomal proteolysis and autophagy require presenilin 1 and are disrupted by Alzheimer-related PS1 mutations. Cell. 2010 Jun 25;141(7):1146-58. PubMed.
Sharma D, Otto G, Warren EC, Beesley P, King JS, Williams RS. Gamma secretase orthologs are required for lysosomal activity and autophagic degradation in Dictyostelium discoideum, independent of PSEN (presenilin) proteolytic function. Autophagy. 2019 Aug;15(8):1407-1418. Epub 2019 Mar 21 PubMed.
Im E, Jiang Y, Lee J-H, Nixon RA. APP-βCTF regulates vATPase-mediated lysosomal acidification. Poster session presented at AD/PD The 14th International Conference on Alzheimer’s & Parkinson’s Diseases. Lisbon, Portugal 2019 March 26-31.
Jiang Y, Sato Y, Im E, Berg M, Bordi M, Darji S, Kumar A, Mohan PS, Bandyopadhyay U, Diaz A, Cuervo AM, Nixon RA. Lysosomal Dysfunction in Down Syndrome Is APP-Dependent and Mediated by APP-βCTF (C99). J Neurosci. 2019 Jul 3;39(27):5255-5268. Epub 2019 May 1 PubMed.
Lee JH, McBrayer MK, Wolfe DM, Haslett LJ, Kumar A, Sato Y, Lie PP, Mohan P, Coffey EE, Kompella U, Mitchell CH, Lloyd-Evans E, Nixon RA. Presenilin 1 Maintains Lysosomal Ca(2+) Homeostasis via TRPML1 by Regulating vATPase-Mediated Lysosome Acidification. Cell Rep. 2015 Sep 1;12(9):1430-44. Epub 2015 Aug 20 PubMed.
Wolfe DM, Lee JH, Kumar A, Lee S, Orenstein SJ, Nixon RA. Autophagy failure in Alzheimer's disease and the role of defective lysosomal acidification. Eur J Neurosci. 2013 Jun;37(12):1949-61. PubMed.
Martín-Maestro P, Gargini R, A Sproul A, García E, Antón LC, Noggle S, Arancio O, Avila J, García-Escudero V. Mitophagy Failure in Fibroblasts and iPSC-Derived Neurons of Alzheimer's Disease-Associated Presenilin 1 Mutation. Front Mol Neurosci. 2017;10:291. Epub 2017 Sep 14 PubMed.
Colacurcio DJ, Nixon RA. Disorders of lysosomal acidification-The emerging role of v-ATPase in aging and neurodegenerative disease. Ageing Res Rev. 2016 May 16; PubMed.
Wallings R, Connor-Robson N, Wade-Martins R. LRRK2 interacts with the vacuolar-type H+-ATPase pump a1 subunit to regulate lysosomal function. Hum Mol Genet. 2019 Aug 15;28(16):2696-2710. PubMed.
Cabeza C, Figueroa A, Lazo OM, Galleguillos C, Pissani C, Klein A, Gonzalez-Billault C, Inestrosa NC, Alvarez AR, Zanlungo S, Bronfman FC. Cholinergic abnormalities, endosomal alterations and up-regulation of nerve growth factor signaling in niemann-pick type C disease. Mol Neurodegener. 2012;7:11. PubMed.
Bagh MB, Peng S, Chandra G, Zhang Z, Singh SP, Pattabiraman N, Liu A, Mukherjee AB. Misrouting of v-ATPase subunit V0a1 dysregulates lysosomal acidification in a neurodegenerative lysosomal storage disease model. Nat Commun. 2017 Mar 7;8:14612. PubMed.
Larner AJ. Presenilin-1 Mutations in Alzheimer's Disease: An Update on Genotype-Phenotype Relationships. J Alzheimers Dis. 2013 Jan 1;37(4):653-9. PubMed.
Make a Comment
To make a comment you must login or register.