Killer Cleavage: Appoptosin Stokes Tauopathy through Caspase 3
Quick Links
Rather than delivering a swift blow to neurons, a death protein found elevated in people with progressive supranuclear palsy (PSP) may exact its toll in a slower, more insidious way. According to a study published September 2 in Neuron, the pro-apoptotic protein appoptosin activates caspase 3, which then cleaves tau. The resulting tau fragments congregated at synapses and damaged them. Animals overexpressing appoptosin displayed movement problems echoing those of people with PSP. The researchers, led by Huaxi Xu at the Sanford Burnham Prebys Medical Discovery Institute in La Jolla, California, propose that this caspase pathway could be at work in other neurodegenerative diseases marked by tauopathy, including Alzheimer’s and some forms of frontotemporal dementia.
PSP is a fatal, age-related neurodegenerative disorder that affects both movement and cognition. The cause of the disease is unknown, although neurofibrillary tau tangles are its main pathological hallmark (see Williams and Lees, 2009). Several single nucleotide polymorphisms (SNPs) in and around the tau gene have been linked to PSP susceptibility. In 2011, a genome-wide association study (GWAS) uncovered a PSP risk locus near an SNP close to the MOBP gene (see Höglinger et al., 2011). However, the researchers found that this SNP did not affect MOBP expression. Rather, its carriers had elevated levels of appoptosin, a gene located 70kb away. How this variant boosts appoptosin levels remains unclear, but Xu told Alzforum that enhanced engagement of promoters or enhancers near the gene likely play a role.
Appoptosin is a mitochondrial protein known to play a part in—you guessed it—apoptosis. It does so by switching on caspase-3, a protease that activates other proteins that execute the death cascade. Xu’s group previously reported that overexpressing appoptosin triggers cell death, while reducing its expression makes cells more resistant to Aβ-mediated toxicity (see Nov 2012 news). Interestingly, tau is a known caspase-3 substrate, and caspase-cleaved tau fragments readily form neurofibrillary tangles (see de Calignon et al., 2010).
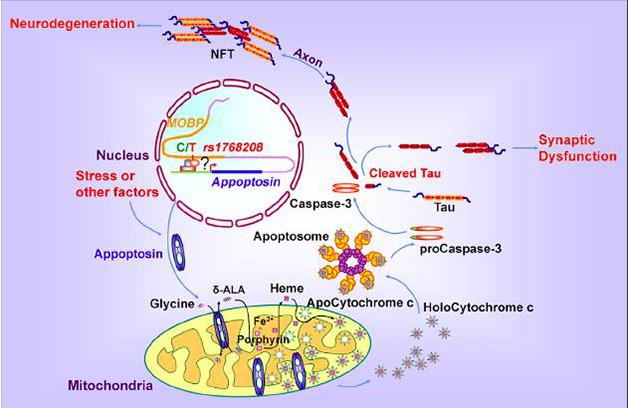
A Slow Death. In this model, increasing appoptosin expression driven by a PSP risk allele triggers a caspase cascade that promotes synaptic dysfunction and neurodegeneration through cleavage of tau. [Courtesy of Zhao et al., Neuron, 2015.]
Could elevated levels of appoptosin mediate tauopathy via caspase-3 in PSP or other disorders? First author Yingjun Zhao and colleagues conducted the current study to find out. They first looked for the disease-associated SNP, and corresponding expression of appoptosin, in postmortem brain samples from PSP patients. Startlingly, they found that 20 of the 26 patients harbored at least one copy of the disease-associated SNP (or T-allele), while the remaining five patients had the C-allele. Only seven out of 22 healthy controls had the T-allele. Expression of appoptosin mRNA and protein was higher in PSP patients than in controls, and those harboring the T-allele had the highest levels.
The researchers found elevated levels of activated caspase-3 as well as its tau cleavage product, c-tau, in PSP brains. Postmortem immunohistochemistry revealed appoptosin, activated caspase-3, c-tau, and paired helical filament (PHF-1) tau comingling in cortical neurons of PSP patients, but not of controls. PSP patients expressing the T-allele had the greatest amount of pathological, PHF-1 tau, suggesting a link between appoptosin expression and the severity of tauopathy.
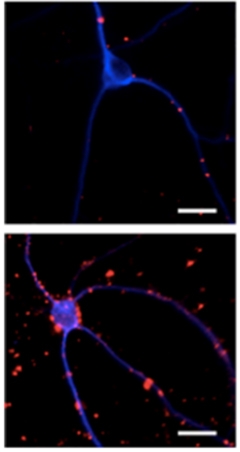
Perilous Puncta?
Clusters of caspase-cleaved tau (red) speckled neurons transduced with appoptosin (bottom panel), but are sparse in neurons transduced with green fluorescent protein only (top). [Courtesy of Zhao et al., Neuron, 2015.]
The researchers next turned to cell culture models to tease out the mechanism of appoptosin-mediated tauopathy. They transduced primary rat cortical neurons with human appoptosin, and found that this overexpression did not trigger cell death until nine days after transduction, despite caspase activation on day two. This delayed death jibes with previous studies suggesting that post-mitotic neurons respond to caspase activation differently than non-neuronal cells (see Hyman and Yuan, 2012). Appoptosin overexpression increased neuronal susceptibility to cell death induced by Aβ or the mitochondrial disrupter MPP. It also boosted levels of c-tau, an effect that was completely abolished by treatment with caspase-3 inhibitors. The researchers concluded that appoptosin-dependent production of c-tau was dependent on caspase-3.
How did appoptosin affect tau trafficking and aggregation? To find out, the researchers first compared exactly where tau and c-tau reside in neurons overexpressing appoptosin or not. They found that appoptosin overexpression led to dissociation of tau from microtubules, and c-tau was found in cell fractions devoid of microtubules. Appoptosin overexpression also increased tau aggregation, as measured by the presence of detergent-insoluble tau in cell extracts. Tau and c-tau parted ways inside the neuron, as immunostaining revealed full-length tau within axons, while c-tau puncta collected in dendrites. Conversely, c-tau puncta were sparse in cells not overexpressing appoptosin, and even fewer in appoptosin-knockout neurons.
Further fractionation studies exposed an abundance of activated caspase-3 and c-tau in synapses. Their presence there had consequences: Neurons overexpressing appoptosin had fewer dendritic spines and expressed fewer neurotransmitter receptors on their surface. Treatment with caspase-3 inhibitors blocked all these effects. Strikingly, in tau-deficient neurons, appoptosin overexpression failed to reduce surface levels of the two receptor subunits, GluR1 and NR1, indicating that caspase-3 has other substrates that influence synapses, as well. These results implicate caspase-3 and, to some extent, c-tau, in synaptic deficits induced by appoptosin.
To test whether appoptosin expression triggers motor deficits in animals, the researchers injected an adeno-associated virus expressing appoptosin directly into the globus pallidus of mice. This small speck of subcortical brain tissue is littered with tau pathology in PSP patients. The mice were JNPL3 transgenics expressing the human tau mutant P301L, which causes some cases of frontotemporal dementia. Compared to JNPL3 mice injected with an empty virus, those transduced with appoptosin walked with shorter strides, and easily lost their balance when placed on a spinning rod or balance beam. The researchers found abundant activated caspase-3, c-tau, and hyperphosphorylated PHF-1 tau near the injection area. Both motor and biochemical effects of appoptosin overexpression were abolished when the researchers infused a caspase-3 inhibitor via minipump. Tau knockout mice transduced with appoptosin developed fewer motor deficits, further implicating tau in the neurodegenerative cascade. Aged JNPL3 mice not transduced with appoptosin also expressed elevated levels of c-tau and PHF-1, suggesting that the aging process itself triggers caspase activation without appoptosin overexpression.
Finally, the researchers compared appoptosin and c-tau in postmortem brain samples from patients with Alzheimer’s, Parkinson’s, and Huntington’s diseases, and FTD with tauopathy. They found elevated levels of both proteins only in the two diseases with apparent tauopathy, namely AD and FTD with tauopathy.
“These exciting findings are a major step forward, since they resolve the previously enigmatic genetic association of variation at the MOBP/SLC25A38 locus with the neurobiology of tauopathies,” wrote Günter Höglinger of the German Center for Neurodegenerative Diseases in Munich and Ulrich Müller of Justus-Liebig-University in Giessen, Germany, in a joint comment to Alzforum. “Furthermore, appoptosin might be the missing link between amyloid-β and tau in Alzheimer’s disease, which has been desperately sought for a long time.”
Xu and colleagues proposed a caspase-driven pathway that could be common to all tauopathies. In the model, appoptosin levels rise, either due to genetic mutations (as in PSP patients harboring the T-allele), or other cellular stresses. Appoptosin then ramps up caspase activity—another factor that could also be independently activated by aging or stress. The caspases cleave tau into c-tau, and the fragments aggregate in synapses, where they cause dysfunction and neurodegeneration. Xu added that caspases likely wreak further havoc by cleaving other substrates as well, although the current study suggests that c-tau plays a pivotal role in the destruction.
Höglinger and Müller also noted that non-genetic factors could activate the pathway in people harboring the non-pathogenic C-allele, which does not upregulate appoptosin. “In patients carrying the C allele, the same pathological mechanism might apply as in patients with the T allele and appoptosin might be activated by environmental factors such as neurotoxins and stress,” they wrote. “It will be interesting to see whether the most severe manifestations of PSP are found among patients carrying the T allele since environmental factors might further increase appoptosin activity.”
Robert Rissman of the University of California, San Diego, described caspase cleavage of tau as a mediator of tau aggregation in AD more than a decade ago (see Rissman et al., 2004). He commented that tau processing has since emerged as a commonality between tauopathies. “When you look at tau as a CSF biomarker for AD, a majority of the protein is cleaved in one way or another,” he said. “The next step in research will be to understand exactly why these cleaved forms are so problematic.”
Rissman added that researchers still have much to learn about the physiological function of tau and its cleavage products, which may go beyond microtubule stabilization. “Gaining a further understanding of the role of tau and its post-translational cleavage will be key to not only understanding the function of tau, but how we can design therapeutics that directly target tau.”
Xu said his lab is currently investigating the role of appoptosin and caspase cleavage of tau in other models of neurodegeneration and brain damage, including traumatic brain injury and stroke, both of which result in tauopathy.—Jessica Shugart
References
News Citations
Research Models Citations
Paper Citations
- Williams DR, Lees AJ. Progressive supranuclear palsy: clinicopathological concepts and diagnostic challenges. Lancet Neurol. 2009 Mar;8(3):270-9. PubMed.
- Höglinger GU, Melhem NM, Dickson DW, Sleiman PM, Wang LS, Klei L, Rademakers R, de Silva R, Litvan I, Riley DE, van Swieten JC, Heutink P, Wszolek ZK, Uitti RJ, Vandrovcova J, Hurtig HI, Gross RG, Maetzler W, Goldwurm S, Tolosa E, Borroni B, Pastor P, PSP Genetics Study Group, Cantwell LB, Han MR, Dillman A, van der Brug MP, Gibbs JR, Cookson MR, Hernandez DG, Singleton AB, Farrer MJ, Yu CE, Golbe LI, Revesz T, Hardy J, Lees AJ, Devlin B, Hakonarson H, Müller U, Schellenberg GD. Identification of common variants influencing risk of the tauopathy progressive supranuclear palsy. Nat Genet. 2011 Jun 19;43(7):699-705. PubMed.
- de Calignon A, Fox LM, Pitstick R, Carlson GA, Bacskai BJ, Spires-Jones TL, Hyman BT. Caspase activation precedes and leads to tangles. Nature. 2010 Apr 22;464(7292):1201-4. PubMed.
- Hyman BT, Yuan J. Apoptotic and non-apoptotic roles of caspases in neuronal physiology and pathophysiology. Nat Rev Neurosci. 2012 May 18;13(6):395-406. PubMed.
- Rissman RA, Poon WW, Blurton-Jones M, Oddo S, Torp R, Vitek MP, Laferla FM, Rohn TT, Cotman CW. Caspase-cleavage of tau is an early event in Alzheimer disease tangle pathology. J Clin Invest. 2004 Jul;114(1):121-30. PubMed.
Further Reading
Papers
- D'Amelio M, Cavallucci V, Middei S, Marchetti C, Pacioni S, Ferri A, Diamantini A, De Zio D, Carrara P, Battistini L, Moreno S, Bacci A, Ammassari-Teule M, Marie H, Cecconi F. Caspase-3 triggers early synaptic dysfunction in a mouse model of Alzheimer's disease. Nat Neurosci. 2011 Jan;14(1):69-76. PubMed.
- Ding H, Matthews TA, Johnson GV. Site-specific phosphorylation and caspase cleavage differentially impact tau-microtubule interactions and tau aggregation. J Biol Chem. 2006 Jul 14;281(28):19107-14. PubMed.
Primary Papers
- Zhao Y, Tseng IC, Heyser CJ, Rockenstein E, Mante M, Adame A, Zheng Q, Huang T, Wang X, Arslan PE, Chakrabarty P, Wu C, Bu G, Mobley WC, Zhang YW, St George-Hyslop P, Masliah E, Fraser P, Xu H. Appoptosin-Mediated Caspase Cleavage of Tau Contributes to Progressive Supranuclear Palsy Pathogenesis. Neuron. 2015 Sep 2;87(5):963-75. PubMed. Correction.
Annotate
To make an annotation you must Login or Register.
Comments
Hannover Medical School
Justus-Liebig University Giessen
Progressive supranuclear palsy (PSP) is a debilitating and deadly neurodegenerative disorder. It is neuropathologically defined by aggregation of the microtubule-associated protein tau in the somata of neurons, oligodendrocytes, and astrocytes. The etiopathogenesis of the disease remains unresolved. Homozygosity for the H1 haplotype of the MAPT gene has been identified as a strong genetic risk factor for PSP. A recent genome-wide association study has confirmed this association and identified three additional risk loci, i.e., STX6, EIF2AK3, and MOBP (Höglinger et al., 2011). The MAPT polymorphism appears to result in altered expression of the protein tau, causing increased aggregation of this protein. In contrast, the pathogenic mechanism of the STX6, EIF2AK3, and MOBP polymorphism has remained unresolved until recently. The association of a genetic polymorphism (T allele of rs1768208) at MOBP (myelin-associated oligodendrocyte basic protein) was particularly puzzling since this gene encodes for an abundantly expressed myelin-constituting protein of the CNS (Yamamoto et al., 1994).
Very interestingly, the rs1768208 MOBP risk polymorphism has also been found to increase risk for the primary tauopathy corticobasal degeneration (CBD, Kouri et al., 2015) and for late-onset Alzheimer’s disease of APOE ε4-positive cases (Liu et al., 2012). Furthermore, the rs1768208 MOBP risk polymorphism has a negative prognostic value in the behavioral-variant frontotemporal dementia. Carriers of the risk (T) allele exhibit more severe white-matter degeneration and shorter disease duration. This particularly applies to autopsy-confirmed patients with tauopathies, but not to cases displaying TDP-43 proteinopathies (Irwin et al., 2014). Taken together, these genetic associations have pointed to a relevant role of the rs1768208 MOBP risk polymorphism in neurodegenerative tauopathies.
In a first attempt to understand the biological mechanism behind this association, the effect of variability at rs1768208 on gene expression in human brains has been studied (Höglinger et al., 2011). SNPs located in or near MOBP showed some moderate effect on MOBP expression. However, a highly significant effect was discovered for SLC25A38 expression, which lies 70 kb from MOBP (Höglinger et al., 2011).
In 2012, Zhang et al. identified the SLC25A38 gene product as a β-amyloid precursor protein (APP)-interacting protein, designated as appoptosin, which is upregulated in brains from patients with Alzheimer's disease. They found appoptosin to be a pro-apoptotic protein that induces caspase-3-dependent apoptosis (Zhang et al., 2012).
Here, the same group reports a high frequency of the T allele and increased levels of the pro-apoptotic protein appoptosin in PSP. They show that increased expression of appoptosin correlates with activated caspase-3 and caspase-cleaved tau levels, tau aggregation, and synaptic dysfunction. In contrast, appoptosin deficiency reduced tau cleavage and aggregation. They also found increased appoptosin and caspase-3-cleaved tau in brains of patients with Alzheimer’s disease and FTLD-Tau.
According to the authors, about 75 percent of PSP patients carry at least one copy of the T allele of rs1768208 and 25 percent do not (they carry the C allele). As the authors convincingly show, the T but not the C allele increases activity of appoptosin, which activates caspase-3, which in turn cleaves tau. In patients carrying the C allele, however, the same pathological mechanism might apply as in patients with the T allele, and appoptosin might be activated by environmental factors such as neurotoxins and stress. It will be interesting to see whether the most severe manifestations of PSP are found among patients carrying the T allele in the homozygous state, since environmental factors might further increase appoptosin activity.
These exciting findings are a major step forward, since they resolve the previously enigmatic genetic association of variation at the MOBP/SLC25A38 locus with the neurobiology of tauopathies. Furthermore, appoptosin might be the missing link between amyloid-β and tau in Alzheimer’s disease, which has been desperately sought for a long time.
This paper is adrenalin! Don’t miss it.
References:
Höglinger GU, Melhem NM, Dickson DW, Sleiman PM, Wang LS, Klei L, Rademakers R, de Silva R, Litvan I, Riley DE, van Swieten JC, Heutink P, Wszolek ZK, Uitti RJ, Vandrovcova J, Hurtig HI, Gross RG, Maetzler W, Goldwurm S, Tolosa E, Borroni B, Pastor P, PSP Genetics Study Group, Cantwell LB, Han MR, Dillman A, van der Brug MP, Gibbs JR, Cookson MR, Hernandez DG, Singleton AB, Farrer MJ, Yu CE, Golbe LI, Revesz T, Hardy J, Lees AJ, Devlin B, Hakonarson H, Müller U, Schellenberg GD. Identification of common variants influencing risk of the tauopathy progressive supranuclear palsy. Nat Genet. 2011 Jun 19;43(7):699-705. PubMed.
Irwin DJ, McMillan CT, Suh E, Powers J, Rascovsky K, Wood EM, Toledo JB, Arnold SE, Lee VM, Van Deerlin VM, Trojanowski JQ, Grossman M. Myelin oligodendrocyte basic protein and prognosis in behavioral-variant frontotemporal dementia. Neurology. 2014 Aug 5;83(6):502-9. Epub 2014 Jul 3 PubMed.
Kouri N, Ross OA, Dombroski B, Younkin CS, Serie DJ, Soto-Ortolaza A, Baker M, Finch NC, Yoon H, Kim J, Fujioka S, McLean CA, Ghetti B, Spina S, Cantwell LB, Farlow MR, Grafman J, Huey ED, Ryung Han M, Beecher S, Geller ET, Kretzschmar HA, Roeber S, Gearing M, Juncos JL, Vonsattel JP, Van Deerlin VM, Grossman M, Hurtig HI, Gross RG, Arnold SE, Trojanowski JQ, Lee VM, Wenning GK, White CL, Höglinger GU, Müller U, Devlin B, Golbe LI, Crook J, Parisi JE, Boeve BF, Josephs KA, Wszolek ZK, Uitti RJ, Graff-Radford NR, Litvan I, Younkin SG, Wang LS, Ertekin-Taner N, Rademakers R, Hakonarsen H, Schellenberg GD, Dickson DW. Genome-wide association study of corticobasal degeneration identifies risk variants shared with progressive supranuclear palsy. Nat Commun. 2015 Jun 16;6:7247. PubMed.
Liu QY, Yu JT, Miao D, Ma XY, Wang HF, Wang W, Tan L. An exploratory study on STX6, MOBP, MAPT, and EIF2AK3 and late-onset Alzheimer's disease. Neurobiol Aging. 2013 May;34(5):1519.e13-7. Epub 2012 Oct 30 PubMed.
Yamamoto Y, Mizuno R, Nishimura T, Ogawa Y, Yoshikawa H, Fujimura H, Adachi E, Kishimoto T, Yanagihara T, Sakoda S. Cloning and expression of myelin-associated oligodendrocytic basic protein. A novel basic protein constituting the central nervous system myelin. J Biol Chem. 1994 Dec 16;269(50):31725-30. PubMed.
Zhang H, Zhang YW, Chen Y, Huang X, Zhou F, Wang W, Xian B, Zhang X, Masliah E, Chen Q, Han JD, Bu G, Reed JC, Liao FF, Chen YG, Xu H. Appoptosin is a novel pro-apoptotic protein and mediates cell death in neurodegeneration. J Neurosci. 2012 Oct 31;32(44):15565-76. PubMed.
Make a Comment
To make a comment you must login or register.