HIPK2 Clinches Neurons’ Fate in Amyotrophic Lateral Sclerosis
Quick Links
Misfolded proteins aggregate … neurons die. What happens in between largely eludes researchers of every neurodegenerative disease, but a new study may have found a piece of the deadly puzzle for amyotrophic lateral sclerosis (ALS). Researchers led by Eric Huang at the University of California, San Francisco, reported that the kinase HIPK2 triggers a death cascade in response to endoplasmic reticulum stress, which is set off by the accumulation of misfolded proteins. The kinase played a role in neurodegeneration in animal models of ALS, and researchers reported elevation of the death cascade in the remaining motor neurons of sporadic and familial ALS patients, but not in people with Alzheimer’s disease. The researchers proposed inhibition of the neuronal henchman as a therapeutic strategy.
“Activation of HIPK2 may be one of the earliest pathogenic events in ALS,” Huang told Alzforum. “Our paper provides proof of concept that this pathway is druggable.”
Accumulating misfolded proteins push all sorts of cellular buttons, one of them being the activation of the unfolded protein response (UPR). This collection of pathways responds to a stressful build-up of proteins in the ER, and can be set off by any of three ER transmembrane proteins: inositol-requiring enzyme-1 (IRE1), PKR-like ER kinase (PERK), or activating transcription factor-6 (ATF6). At first, the UPR attempts to clean up the mess by ramping up chaperone expression and degradation pathways, but if the stress persists, the response switches into suicide mode and triggers cell death instead (Oakes and Papa, 2015). People with sporadic ALS have signs of ER stress in their motor neurons (Atkin et al., 2008). Familial forms of the disease—whether caused by hexanucleotide expansions in the C9ORF72 gene or mutations in TAR DNA binding protein 43 (TDP-43), superoxide dismutase (SOD1), or fused in sarcoma (FUS)—are all characterized by problems in RNA metabolism and protein homeostasis, which could trip off ER stress (Walker and Atkin, 2011; Ling et al., 2013). In mouse models of mutant SOD1, researchers have reported that the kinase PERK is activated and prolongs survival (Wang et al., 2011). However, little is known about how a different branch of the ER stress response—marked by the activation of the kinase IRE1α—affects the fate of neurons.
Huang and colleagues discovered by serendipity that HIPK2—a kinase they had implicated more than a decade ago in developmental cell death in neurons—was playing a more sinister role in this branch of the UPR pathway in ALS (Wiggins et al., 2004; Zhang et al., 2007).
First author Sebum Lee and colleagues started by treating embryonic fibroblasts from normal or HIPK2 knockout mice with a variety of inhibitors known to induce cell death. Both wild-type and knockout cells were equally vulnerable to protein synthesis inhibitors, but HIPK2 knockout cells gamely resisted tunicamycin, a bacterial toxin that promotes accumulation of unglycosylated proteins in the ER. Cultured neurons from HIPK2 knockout mice were resistant, too, and knocking down HIPK2 in HEK293 cells helped those cells withstand tunicamycin, as well.
The researchers next moved their investigation in vivo by injecting mice with tunicamycin and monitoring what it did to neurons four days later. In wild-type mice, the treatment killed nearly a third of cortical neurons and ramped up markers of cell death in the brain, yet the number of cortical neurons in HIPK2 knockout mice remained unchanged. Spinal motor neurons in these mice also resisted tunicamycin’s toxic effects.
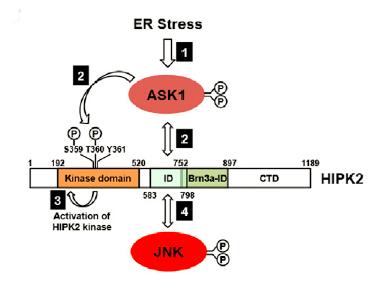
Asking for Death.
ER stress triggers a neuronal death cascade by activating ASK1, which phosphorylates HIPK2, which then becomes activated and phosphorylates JNK. [Courtesy of Lee et al., Neuron 2016.]
With further experiments, the researchers pieced together the signaling cascade that brought about neuronal death. They found that misfolded proteins activated the IRE1α wing of the unfolded protein response, which triggered activation of apoptosis-signal regulating kinase 1. ASK1 then phosphorylated HIPK2 on two essential residues—threonine-360 and serine-359. Thus activated, HIPK2 then formed a complex with JNK, which upregulated apoptotic genes that executed the neuron’s demise.
Pathway in hand, the researchers looked for activation of the death cascade in animal models of ALS. In the prominent SOD1-G93A mutant mice, HIPK2 expression and phosphorylation appeared prior to disease onset and increased with age in spinal motor neurons. In the regulatable NLS mice expressing a mutant form of TDP-43 that can be induced by stopping dietary doxycycline, the researchers observed elevated levels of phosphorylated IRE1α, HIPK2, and JNK 11 weeks after disease induction. In contrast, motor neurons in FUS-R521C mice did not appear to activate the pathway. Huang assumes this is because few neurons actually die in FUS-R521C mice, as opposed to the rampant death observed in SOD1 and TDP-43 models.
The researchers next deleted HIPK2 from SOD1-G93A mice, finding that the crosses got sick at an older age, were more coordinated, and lived longer than mice expressing the kinase. The researchers also reported that downstream JNK activation decreased by half in SOD1-G93A mice expressing one copy of HIPK2, and by 80 percent in mice lacking HIPK2. This pegged HIPK2 as the primary activator of JNK in spinal motor neurons in ALS mice. Together, the findings suggested that HIPK2 promoted neurodegeneration, at least in the SOD1-G93A mouse model.

HIP Henchman.
Phosphorylated c-Jun (red, top) and caspase 3 (red, bottom) in confocal micrographs of spinal motor neurons (green) from wildtype (left), SOD1 transgenic mice with HIPK2 (middle), and SOD1 tg without HIPK2 expression (right). [Courtesy of Lee et al., Neuron 2016.]
What about HIPK2 in ALS? Huang gathered samples from UCSF and collaborated with Virginia Lee and John Trojanowski at the University of Pennsylvania in Philadelphia, along with Eileen Bigio at Northwestern University in Chicago, to compile postmortem samples from more than 50 people with ALS. In western blots of spinal cord extracts from three patients harboring one of three different SOD1 mutations, the researchers detected elevated IRE1α, HIPK2, and JNK phosphorylation. They used two-color staining to reveal a twofold increase in HIPK2 staining in five other patients with different SOD1 mutations. The researchers also prepared spinal cord lysates from 28 cases of sporadic ALS and 16 cases of C9ORF72 ALS, as well as 13 controls. They found elevated phosphorylated HIPK2 in most of the sporadic and C9ORF72 cases, although the levels varied widely between people. Importantly, the level of phosphorylated HIPK2 correlated with that of ubiquitinated and phosphorylated TDP-43. In contrast, a similar analysis of frontal cortex samples derived from Alzheimer’s patients or controls revealed no activation of the pathway.
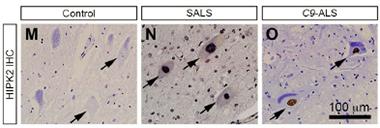
Human HIP.
HIPK2 expression was elevated in samples from people with sporadic ALS (middle) or C9-ALS (right). [Courtesy of Lee et al., Neuron 2016.]
Huang believes that blocking HIPK2 activation potentially could be therapeutic. To test this idea, the researchers applied the kinase inhibitor A64 to spinal motor neurons from SOD1-G93A mice, to neurons poisoned with tunicamycin, to HEK293 cells, and to HEK293 cells or neurons expressing a range of TDP-43 mutations associated with ALS. In each case, treatment blocked HIPK2 phosphorylation and rescued cells from death.
Raymond Roos of the University of Chicago found the data provocative but would like to see larger numbers of animals in the survival experiments. He suggested the study expand to include more ALS models, such as newly available C9ORF72 mice. Roos’ previous work reported that PERK, a kinase activated in another branch of the UPR pathway, promoted neuronal survival in ALS. “What is important here is that the UPR can be very neuroprotective, but also can lead to apoptosis when overwhelmed by misfolded proteins,” Roos said. He added that therefore the timing of any intervention could be crucial. “It could be that enhancing the UPR is beneficial early on, but once there is an overwhelming amount of misfolded protein, then perhaps disabling the UPR or knocking down the apoptotic downstream pathway is important.”
Jonathan Lin of the University of California, San Diego, agreed, adding that both types of therapy could be delivered in combination. “Because this system both promotes survival and death, it offers the possibility for twofold therapies,” he said. A combination treatment could activate PERK and block HIPK2 at the same time. Lin added that, being a kinase, HIPK2 makes an excellent druggable target. Other proteins implicated in ALS are not enzymes and therefore more elusive targets.
Huang wants to collaborate with industry to develop HIPK2 kinase inhibitors that cross the blood-brain barrier, as the one used in his study does not. He added that because ALS patients are remarkably heterogeneous in terms of activation of the HIPK2 death cascade, it would be important to develop a screening tool to stratify patients who would be most likely to respond to treatment. Detection methods to measure phosphorylated HIPK2 released from degenerating neurons in the cerebrospinal fluid would be a key step. Because HIPK2 activation correlated with TDP-43 pathology, such sensors could also be biomarker for disease severity or progression, Huang said.—Jessica Shugart
References
Research Models Citations
Paper Citations
- Oakes SA, Papa FR. The role of endoplasmic reticulum stress in human pathology. Annu Rev Pathol. 2015;10:173-94. Epub 2014 Oct 27 PubMed.
- Atkin JD, Farg MA, Walker AK, McLean C, Tomas D, Horne MK. Endoplasmic reticulum stress and induction of the unfolded protein response in human sporadic amyotrophic lateral sclerosis. Neurobiol Dis. 2008 Jun;30(3):400-7. PubMed.
- Walker AK, Atkin JD. Stress signaling from the endoplasmic reticulum: A central player in the pathogenesis of amyotrophic lateral sclerosis. IUBMB Life. 2011 Aug 10; PubMed.
- Ling SC, Polymenidou M, Cleveland DW. Converging mechanisms in ALS and FTD: disrupted RNA and protein homeostasis. Neuron. 2013 Aug 7;79(3):416-38. PubMed.
- Wang L, Popko B, Roos RP. The unfolded protein response in familial amyotrophic lateral sclerosis. Hum Mol Genet. 2011 Mar 1;20(5):1008-15. PubMed.
- Wiggins AK, Wei G, Doxakis E, Wong C, Tang AA, Zang K, Luo EJ, Neve RL, Reichardt LF, Huang EJ. Interaction of Brn3a and HIPK2 mediates transcriptional repression of sensory neuron survival. J Cell Biol. 2004 Oct 25;167(2):257-67. Epub 2004 Oct 18 PubMed.
- Zhang J, Pho V, Bonasera SJ, Holtzman J, Tang AT, Hellmuth J, Tang S, Janak PH, Tecott LH, Huang EJ. Essential function of HIPK2 in TGFbeta-dependent survival of midbrain dopamine neurons. Nat Neurosci. 2007 Jan;10(1):77-86. Epub 2006 Dec 10 PubMed.
Further Reading
Papers
- Hiramatsu N, Chiang WC, Kurt TD, Sigurdson CJ, Lin JH. Multiple Mechanisms of Unfolded Protein Response-Induced Cell Death. Am J Pathol. 2015 Jul;185(7):1800-8. Epub 2015 May 5 PubMed.
Primary Papers
- Lee S, Shang Y, Redmond SA, Urisman A, Tang AA, Li KH, Burlingame AL, Pak RA, Jovičić A, Gitler AD, Wang J, Gray NS, Seeley WW, Siddique T, Bigio EH, Lee VM, Trojanowski JQ, Chan JR, Huang EJ. Activation of HIPK2 Promotes ER Stress-Mediated Neurodegeneration in Amyotrophic Lateral Sclerosis. Neuron. 2016 Jul 6;91(1):41-55. Epub 2016 Jun 16 PubMed.
Annotate
To make an annotation you must Login or Register.
Comments
No Available Comments
Make a Comment
To make a comment you must login or register.