Genetics Tie ALS into the Frontotemporal Dementia Spectrum
Quick Links
How genetically similar is amyotrophic lateral sclerosis to other neurodegenerative diseases? Researchers led by Celeste Karch, Washington University, St. Louis, and Rahul Desikan, University of California, San Francisco, used genome-wide association data from nearly 125,000 individuals to search for commonalities with six other neurodegenerative diseases. They found a robust overlap between ALS and frontotemporal dementias, including corticobasal degeneration and progressive supranuclear palsy, but not between ALS and Parkinson’s or ALS and Alzheimer’s disease. The study also fingered new ALS risk factors: the H1 haplotype of the tau gene, a known risk factor for several neurodegenerative diseases, and BNIP1, which codes for a multifunctional protein suppressed in spinal cords of ALS patients. The results appear in the April 9 JAMA Neurology.
- A meta-GWAS finds ALS shares risk factors with diseases of the FTD spectrum.
- No shared variants were found in ALS and Parkinson’s or ALS and Alzheimer’s disease.
- The tau H1 haplotype and the BNIP1 gene emerged as new ALS risk factors.
“The study presents several interesting new findings: the association of MAPT H1 with ALS risk, the discovery of new polymorphisms in the C9ORF72 locus, and the new BNIP1 risk gene,” said Julie van der Zee of the VIB-UAntwerp Center for Molecular Neurology, Belgium.
Karch, Desikan and colleagues looked for genetic commonalities among ALS, Alzheimer’s disease, Parkinson’s disease, corticobasal degeneration (CBD), progressive supranuclear palsy (PSP), sporadic frontotemporal dementia (FTD), and autosomal dominant FTD with TDP-43 inclusions. They used data from prior GWAS's, including those run by the International FTD Genomics Consortium, the International Collaboration for FTD, the PSP Genetics Consortium, and the International PD Genomics Consortium. Altogether, the authors analyzed data from 124,876 cases and controls. The ALS data set comprised 31 independent cohorts, including 12,577 patients and 23,475 controls (Van Rheenen et al., 2016).
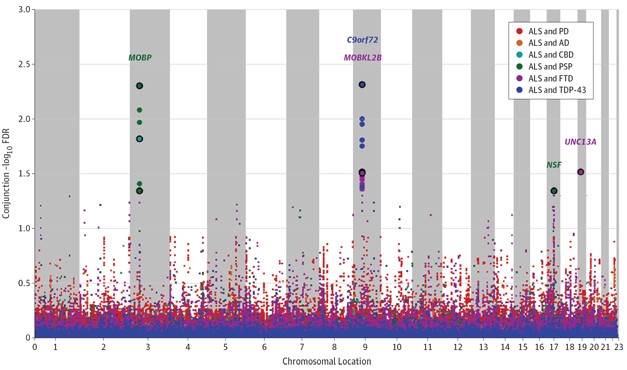
Shared Risk. A genome-wide analysis identified risk variants (colored dots) shared by ALS and any one of six other neurodegenerative disorders. Large points represent significant associations. Genes nearest the most robustly associated SNPS (circled points) are named. [Courtesy of Karch et al., JAMA Neurol, 2018.]
The researchers fished out shared loci pairwise, calculating the likelihood that a given single nucleotide polymorphism (SNP) associates with ALS and any of the other diseases. They found 150-fold and 300-fold enrichments of ALS-associated SNPs in sporadic FTD and in sporadic FTD with TDP-43 inclusions, respectively. Five SNPs emerged as linked to an increased risk for having both ALS and a disease on the FTD spectrum, including SNPs near the MOBP, C9ORF72, MOBKL2B, NSF, and UNC13A genes. ALS SNPs enriched the CBD and PSP data sets 25- and 75-fold, respectively.
The researchers found no commonalities between ALS and AD or ALS and PD. “We knew before that familial forms of FTD and ALS share rare mutations, but here we are seeing [shared] common variants that could help explain sporadic disease,” said Karch. She thinks other shared variants may emerge if larger data sets are explored. “What we are seeing here are only the stronger drivers of shared risk,” she said.
The authors also searched the data for new ALS risk loci. Their idea was that variants that failed to reach significance in the original ALS GWAS might rise above the noise when data were combined. This analysis identified 29 ALS risk loci, 22 of which were new. That number seemed low to Desikan. “To me, it is striking that in terms of polygenic pleiotropy ALS appears very different from AD, PD, and FTD” he wrote. “In comparison to these more common diseases, there is a relative paucity of common variants associated with ALS, which is unlikely due to sample size.”
The new SNPs include several near C9ORF72, and one in the H1 haplotype containing the tau gene. “We don’t usually think of tau involvement in ALS, but one can imagine it could be linked to axonal transport deficits known to occur in ALS,” said Karch. Indeed, a recent paper reported slower axonal transport in neurons derived from healthy MAPTH1 carriers (Beevers et al., 2017).
To dissect potential effects of the shared SNPs, the authors looked for expression quantitative trait loci that associated with them. One, rs7224296, turned out to strongly modulate expression of KIAA1267, a protein involved in histone acetylation, and it co-inherits with rs199533, a known risk variant for PSP, CBD, and FTD (Yokoyama et al., 2017). This SNP, which also tags the MAPTH1 haplotype, modulates expression and splicing of tau to favor the 2N isoform, which contains two N-terminal inserts. SNPs rs13302855 and rs3849942 near C9ORF72 were modestly associated with changes in expression of the LRRC19 and MOBKL2B genes, respectively. These distinct effects on expression, and the separate inheritance of the two SNPs, suggest two independent risk factors residing in the same C9ORF72 locus. Another C9ORF72 SNP, rs2282241, associated with retention of an intron in the C9ORF72 transcript.
One new ALS variant, rs538622, correlated with reduced expression of BNIP1, a gene not previously linked to either ALS or FTD. BNIP1 encodes a protein that appears to block apoptotic suppressors and helps maintain the endoplasmic reticulum network. One study reports that it binds to the mitochondrial ubiquitin ligase RNF185 (Tang et al., 2011), leading the authors to speculate that rs538622 could disrupt the needed disposal of unwanted mitochondria. Two other ALS genes, optineurin and TBK1, have been linked to stalled mitophagy (Sep 2015 news).
However, the human nervous system does not seem to express RNF185, and the functional consequences of its binding to BNIP1 are unexplored. “Finding BNIP is a shared genetic risk factor is interesting, but the biology is unclear at this point,” wrote Giovanni Manfredi of Cornell University, New York, in an email to Alzforum. Karch agreed, and said her lab plans to examine BNIP1 mechanisms in cellular and animal models of ALS. Hints from prior studies suggest ALS spinal cords have half as much BNIP1 as does control tissue (Dangond et al., 2004). Spinal cords of SOD1-G93A mouse models of ALS express about 20 percent less BNIP1 than do wild-type cords (Lukas et al., 2006).—Marina Chicurel
References
News Citations
Research Models Citations
Paper Citations
- van Rheenen W, Shatunov A, Dekker AM, McLaughlin RL, Diekstra FP, Pulit SL, van der Spek RA, Võsa U, de Jong S, Robinson MR, Yang J, Fogh I, van Doormaal PT, Tazelaar GH, Koppers M, Blokhuis AM, Sproviero W, Jones AR, Kenna KP, van Eijk KR, Harschnitz O, Schellevis RD, Brands WJ, Medic J, Menelaou A, Vajda A, Ticozzi N, Lin K, Rogelj B, Vrabec K, Ravnik-Glavač M, Koritnik B, Zidar J, Leonardis L, Grošelj LD, Millecamps S, Salachas F, Meininger V, de Carvalho M, Pinto S, Mora JS, Rojas-García R, Polak M, Chandran S, Colville S, Swingler R, Morrison KE, Shaw PJ, Hardy J, Orrell RW, Pittman A, Sidle K, Fratta P, Malaspina A, Topp S, Petri S, Abdulla S, Drepper C, Sendtner M, Meyer T, Ophoff RA, Staats KA, Wiedau-Pazos M, Lomen-Hoerth C, Van Deerlin VM, Trojanowski JQ, Elman L, McCluskey L, Basak AN, Tunca C, Hamzeiy H, Parman Y, Meitinger T, Lichtner P, Radivojkov-Blagojevic M, Andres CR, Maurel C, Bensimon G, Landwehrmeyer B, Brice A, Payan CA, Saker-Delye S, Dürr A, Wood NW, Tittmann L, Lieb W, Franke A, Rietschel M, Cichon S, Nöthen MM, Amouyel P, Tzourio C, Dartigues JF, Uitterlinden AG, Rivadeneira F, Estrada K, Hofman A, Curtis C, Blauw HM, van der Kooi AJ, de Visser M, Goris A, Weber M, Shaw CE, Smith BN, Pansarasa O, Cereda C, Del Bo R, Comi GP, D'Alfonso S, Bertolin C, Sorarù G, Mazzini L, Pensato V, Gellera C, Tiloca C, Ratti A, Calvo A, Moglia C, Brunetti M, Arcuti S, Capozzo R, Zecca C, Lunetta C, Penco S, Riva N, Padovani A, Filosto M, Muller B, Stuit RJ, PARALS Registry, SLALOM Group, SLAP Registry, FALS Sequencing Consortium, SLAGEN Consortium, NNIPPS Study Group, Blair I, Zhang K, McCann EP, Fifita JA, Nicholson GA, Rowe DB, Pamphlett R, Kiernan MC, Grosskreutz J, Witte OW, Ringer T, Prell T, Stubendorff B, Kurth I, Hübner CA, Leigh PN, Casale F, Chio A, Beghi E, Pupillo E, Tortelli R, Logroscino G, Powell J, Ludolph AC, Weishaupt JH, Robberecht W, Van Damme P, Franke L, Pers TH, Brown RH, Glass JD, Landers JE, Hardiman O, Andersen PM, Corcia P, Vourc'h P, Silani V, Wray NR, Visscher PM, de Bakker PI, van Es MA, Pasterkamp RJ, Lewis CM, Breen G, Al-Chalabi A, van den Berg LH, Veldink JH. Genome-wide association analyses identify new risk variants and the genetic architecture of amyotrophic lateral sclerosis. Nat Genet. 2016 Sep;48(9):1043-8. Epub 2016 Jul 25 PubMed.
- Beevers JE, Lai MC, Collins E, Booth HD, Zambon F, Parkkinen L, Vowles J, Cowley SA, Wade-Martins R, Caffrey TM. MAPT Genetic Variation and Neuronal Maturity Alter Isoform Expression Affecting Axonal Transport in iPSC-Derived Dopamine Neurons. Stem Cell Reports. 2017 Aug 8;9(2):587-599. Epub 2017 Jul 6 PubMed.
- Yokoyama JS, Karch CM, Fan CC, Bonham LW, Kouri N, Ross OA, Rademakers R, Kim J, Wang Y, Höglinger GU, Müller U, Ferrari R, Hardy J, International FTD-Genomics Consortium (IFGC), Momeni P, Sugrue LP, Hess CP, James Barkovich A, Boxer AL, Seeley WW, Rabinovici GD, Rosen HJ, Miller BL, Schmansky NJ, Fischl B, Hyman BT, Dickson DW, Schellenberg GD, Andreassen OA, Dale AM, Desikan RS. Shared genetic risk between corticobasal degeneration, progressive supranuclear palsy, and frontotemporal dementia. Acta Neuropathol. 2017 May;133(5):825-837. Epub 2017 Mar 7 PubMed.
- Tang F, Wang B, Li N, Wu Y, Jia J, Suo T, Chen Q, Liu YJ, Tang J. RNF185, a novel mitochondrial ubiquitin E3 ligase, regulates autophagy through interaction with BNIP1. PLoS One. 2011;6(9):e24367. Epub 2011 Sep 9 PubMed.
- Dangond F, Hwang D, Camelo S, Pasinelli P, Frosch MP, Stephanopoulos G, Brown RH, Gullans SR. Molecular signature of late-stage human ALS revealed by expression profiling of postmortem spinal cord gray matter. Physiol Genomics. 2004 Jan 15;16(2):229-39. PubMed.
- Lukas TJ, Luo WW, Mao H, Cole N, Siddique T. Informatics-assisted protein profiling in a transgenic mouse model of amyotrophic lateral sclerosis. Mol Cell Proteomics. 2006 Jul;5(7):1233-44. PubMed.
Further Reading
Primary Papers
- Karch CM, Wen N, Fan CC, Yokoyama JS, Kouri N, Ross OA, Höglinger G, Müller U, Ferrari R, Hardy J, Schellenberg GD, Sleiman PM, Momeni P, Hess CP, Miller BL, Sharma M, Van Deerlin V, Smeland OB, Andreassen OA, Dale AM, Desikan RS, International Frontotemporal Dementia (FTD)–Genomics Consortium, International Collaboration for Frontotemporal Dementia, Progressive Supranuclear Palsy (PSP) Genetics Consortium, and International Parkinson’s Disease Genomics Consortium. Selective Genetic Overlap Between Amyotrophic Lateral Sclerosis and Diseases of the Frontotemporal Dementia Spectrum. JAMA Neurol. 2018 Jul 1;75(7):860-875. PubMed.
Annotate
To make an annotation you must Login or Register.
Comments
No Available Comments
Make a Comment
To make a comment you must login or register.