GATA4 and Cellular Senescence Said to Stoke ‘Inflammaging’
Quick Links
When one machine in an assembly line goes on the fritz, sometimes the only viable option is to shut down the whole factory. DNA damage can trigger a similar response in cells, called senescence. As described in Science on September 25, researchers led by Stephen Elledge at Brigham and Women’s Hospital in Boston pieced together several key players in the cellular shut-down sequence. They found that the DNA damage response allowed GATA4, a transcription factor, to evade its physiological degradation by autophagy. GATA4 then turned on a slew of inflammatory genes and promoted cellular senescence. The discovery unveils a new wing of the DNA damage response that not only halts cell division, but also could create a damaging inflammatory environment. The researchers found that GATA4 levels in human brains crept up with age, but whether levels of the protein correlate with neuroinflammation and neurodegenerative disease remains to be tested.
Cellular senescence serves to stop the propagation of abnormal cells by turning off cell division. Multiple signals can trigger senescence. They include DNA damage, shortened telomeres, replicative exhaustion, and expression of oncogenes, all of which increase with age (see Kuilman et al., 2010; Campisi and Robert, 2014; Rossiello et al., 2014; Khurana and Oberdoerffer, 2015). Although a senescent cell's heyday of proliferation may be over, it is by no means dormant. Drastic changes in gene expression accompany senescence, including a rise in pro-inflammatory cytokines and chemokines, growth factors, and proteases. Called the senescence-associated secretory phenotype (SASP), this state of pro-inflammatory secretion can help maintain a cell’s state of senescence or influence nearby cells to do the same (see Coppé et al., 2010).
Researchers propose that SASP-stricken cells contribute to the chronic inflammation observed in many age-related diseases, but the mechanisms that switch on this program in response to damage have eluded researchers (see Ovadya and Krizhanovsky, 2014; Zhu et al., 2014). Autophagy—a process by which the cell disposes of its own contents within lysosomes—plays a controversial role in senescence. Some studies indicate that autophagy prevents senescence, while others report it brings it on (see Gewirtz 2013).
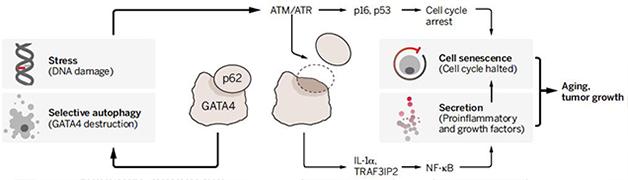
Inflammatory Branch. In this model, DNA damage releases GATA4 from disposal by autophagy, so it can switch on pro-inflammatory genes. Meanwhile, a separate pathway also mediates senescence. [Courtesy of V. Altounian, Science 2015.]
To elucidate the switches that regulate senescence, first author Chanhee Kang and colleagues first looked for microRNAs that were specifically turned on in senescent cells. miR146 fit the bill, and GATA4 transcription factor was required to turn on its expression. GATA4 is a zinc finger transcription factor that belongs to the larger GATA family, and is known to orchestrate the development of several organs.
This initial link between GATA4 and the expression of a senescence-specific microRNA led the researchers to probe deeper into the role GATA4 plays in the broader pathways of senescence. When they overexpressed GATA4 in human foreskin fibroblasts, the cells entered a senescent state, as measured by an increase in β-galactosidase activity that occurs when cells senesce (SA-β-Gal). Conversely, when the researchers depleted the cells of GATA4 using short hairpin RNAs, or mutated it using CRISPR, they found that the fraction of cells entering a senescent state after being zapped with ionizing radiation dropped by more than half. This data indicated that GATA4 was a key player in senescence.
The amount of GATA4 protein, but not mRNA, dramatically increased in fibroblasts following induction of senescence. To find out how GATA4 was regulated at the protein level, the researchers treated cells with inhibitors of classical protein degradation pathways—the proteasome and the lysosome. They found that lysosomal, but not proteosomal, degradation was responsible for the normally low levels of GATA4 in non-senescent cells. Specifically, they found that depletion of the autophagy factors ATG5 or ATG7 boosted GATA4 expression.
GATA4 levels also shot up when the researchers deprived cells of p62, a protein that funnels proteins into autophagy. This result hinted that in non-senescent cells, p62 normally targets GATA4 to a selective autophagy pathway. P62 is no stranger to neurodegenerative disease: mutations in its gene have been linked to frontotemporal dementia and amyotrophic lateral sclerosis (ALS), where the protein crops up in aggregates (see Nov 2011 news; and Sep 2015 news).
The researchers proposed that senescence-inducing signals such as ionizing radiation could release GATA4 from this self-limiting destruction shuttle, thus allowing it to promote senescence. If so, this could explain conflicting conclusions in previous studies about the role of autophagy in senescence. Perhaps only selective autophagy of certain substrates (such as GATA4) prevented senescence, while general autophagy helped promote senescence later on? In keeping with this idea, the researchers found that transiently switching off autophagy more effectively triggered senescence than shutting it down permanently; depleting cells of p62 induced senescence even more.
Once freed from the chains of autophagy, how did GATA4 drive senescence? Because it is a known transcriptional activator, the researchers used RNA sequencing to look for changes in gene expression following GATA4 induction in fibroblasts. They found many SASP-associated genes upregulated, including inflammatory cytokines such as IL6 and chemokines such as CXCL1, while genes that promote progression through the cell cycle were turned off. Interestingly, a significant portion of the genes that switched on in response to GATA4 were the same as those triggered when senescence was induced. By depleting cells of the nuclear factor κB subunit RELA, the researchers found that GATA4 upregulated many SASP genes by way of NFκB. Drilling down even further, they found that GATA4’s induction of two genes—TRAF3IP2 and IL1α—activated NFκB, which then switched on many of the SASP genes.
Collectively, these findings indicated that in response to stressors, GATA4 stabilizes and triggers the induction of SASP genes through the activation of NFκB. The researchers proposed that GATA4 was responsible for much of the inflammatory response that occurs in senescent cells, and that this response may then further fuel senescence.
The next obvious question was: What signals initially trigger GATA4’s release from the clutches of autophagy? As most senescence-inducing signals involve DNA damage, the researchers tested whether the DNA damage response (DDR) was necessary for GATA4 stabilization and subsequent SASP. They treated fibroblasts with inhibitors of the DDR activators ATM and ATR (ataxia telangiectasia mutated or Rad3-related), and found that the treated cells failed to stabilize GATA4 or activate NFκB in response to ionizing radiation. This indicated that the DDR triggers the stabilization of GATA4 and subsequent expression of SASP genes. It is still unclear how the DDR releases GATA4 from p62 and autophagy, Elledge said. “It could be that GATA4 is modified so it can’t be targeted, or that p62 is prevented from binding substrates, including GATA4,” he said.
This GATA4 pathway ran in parallel to another, established DDR-mediated pathway to senescence. It involves p53 and p16INK4a. The researchers found that these factors also use ATM and ATR to plunge cells into a senescent state. However, the separate GATA4 pathway was required to also turn on SASP genes. Elledge proposed that these gene expression changes triggered by GATA4 serve to reinforce the senescent state.
“A complex picture has emerged in which the major upstream regulators of senescence and the SASP have been described in isolation,” wrote Liam Cassidy and Masashi Narita of the University of Cambridge in England, in an accompanying commentary. “Each is associated with a multitude of diverse effects, downstream signaling, and complex feedback loops, perhaps providing a dynamic fine-tuning mechanism for the SASP regulatory network.”

Go Go GATA.
DNA damage ramps up GATA4 expression in mouse embryonic fibroblasts (lower right). [Courtesy of Kang et al., Science 2015.]
Findings in cultured fibroblasts are well and good, but would this pathway unfold in aging humans? To approach this leap, the researchers first exposed grown mice to radiation, which is known to trigger senescence in organs. That boosted amounts of GATA4 in the skin and liver. Aging also switched on the pathway, as evidenced by elevated levels of GATA4 and NFκB activation in the livers and kidneys of 22-month-old mice compared to 6-month-olds. The same appeared to be true in the human brain. The researchers compared levels of GATA4 and another senescence marker, p16INK4a, in the prefrontal cortex of postmortem brain samples from three people in their mid-20s to those of five people in their 70s to 90s without a diagnosis of AD or any other neurodegenerative disease. They found that both proteins were elevated in old people, where they comingled in neurons, oligodendrocytes, and astrocytes.
Elledge proposed that GATA4 could be a useful indicator of accumulating damage associated with aging, and also a sign that potentially harmful inflammatory responses are on the rise. While he plans to unravel the molecular web that links DNA damage to the stabilization of GATA4, much is left to be explored by other researchers in regard to how GATA4-mediated inflammation and senescence tie in with aging, Elledge told Alzforum. Interestingly, while GATA4 rises with age, a previous study from co-author Bruce Yankner’s lab at Harvard Medical School in Boston reported that another transcription factor, REST, plummets. REST is targeted by autophagy. Low levels of REST made cells more vulnerable to cellular stressors such as oxidative stress, and low levels of the protein correlated with cognitive decline (see Mar 2014 news). Connections between these yin and yang transcription factors have yet to be made. “There is no question that genomic instability and DNA damage increase in the brain with aging,” said Yankner.
At the same time, both systemic and neuroinflammation stoke trouble as well, in a process sometimes referred to as “inflammaging” (see Franceschi and Campisi, 2014; Giunta et al., 2008). Whether the GATA4-mediated SASP response is the link between these two phenomena is still unclear, Yankner said. Which cells in the brain predominantly manifest the SASP response also remains unknown. Researchers need to address whether boosted GATA4 levels trigger neurons to secrete inflammatory proteins, which is normally a job reserved for glial cells, Yankner added.
Lastly, researchers have yet to define the role of GATA4 in neurodegenerative disease. Some researchers have proposed that environmental stressors switch on senescence and SASP in glial cells, which promotes Parkinson’s disease (see Chinta et al., 2013). Previous studies implicated other GATA family members in the production of α-synuclein, but this was unrelated to the SASP pathway switched on by GATA4 (see Jul 2008 news). Researchers have yet to understand whether people with AD or other disorders harbor higher levels of GATA4, and if so, whether this contributes to or is just a consequence of disease.
While the GATA4 connection is new, researchers have previously linked NFκB activation to neurodegenerative disease. For example, Jean-Pierre Julien of Laval University in Montreal reported postmortem evidence of elevated levels of the activated factor in temporal lobe neurons of people with MCI, as well as in motor neurons from people with ALS (Ohta et al., 2014; Dec 2011 news). Age-related NFκB activation emerged in mouse models at the same time that toxic aggregates cropped up. Julien wrote to Alzforum that it would be interesting to see if GATA4 went up at the same time (see comment below).
Mark Albers of Massachusetts General Hospital, who was not involved in the study, commented that the findings revealed an interesting link between DNA damage and neuroinflammation, though the details of the pathway still need to be ironed out. “These findings have important translational potential,” he added. “Preventing or reversing the accumulation of GATA4 in the aging brain may offer therapeutic benefit by reducing the burden of neuroinflammation and thereby reducing the clinical manifestations of aging and neurodegenerative disease.”
Would GATA4 itself make a good drug target? Elledge thinks not, because as a transcription factor it targets many genes. Moreover, its known role in development could extend into adulthood, especially in the heart and other tissues that renew themselves.—Jessica Shugart
References
News Citations
- DC: Protein Work Expands ALS/FTD Genetics
- Rare FTD Cases Another Example of Lysosomal Pathology in Dementia
- No REST for Weary Neurons: Protective Factor Stems Cognitive Decline
- A PD Trifecta: Synuclein Gene Expression, Aging, and Inflammation
- TDP-43 Hangs Out With NF-κB, Puts Innate Immunity Into Hyperdrive
Paper Citations
- Kuilman T, Michaloglou C, Mooi WJ, Peeper DS. The essence of senescence. Genes Dev. 2010 Nov 15;24(22):2463-79. PubMed.
- Campisi J, Robert L. Cell senescence: role in aging and age-related diseases. Interdiscip Top Gerontol. 2014;39:45-61. Epub 2014 May 13 PubMed.
- Rossiello F, Herbig U, Longhese MP, Fumagalli M, d'Adda di Fagagna F. Irreparable telomeric DNA damage and persistent DDR signalling as a shared causative mechanism of cellular senescence and ageing. Curr Opin Genet Dev. 2014 Jun;26:89-95. Epub 2014 Aug 11 PubMed.
- Khurana S, Oberdoerffer P. Replication Stress: A Lifetime of Epigenetic Change. Genes (Basel). 2015 Sep 11;6(3):858-77. PubMed.
- Coppé JP, Desprez PY, Krtolica A, Campisi J. The senescence-associated secretory phenotype: the dark side of tumor suppression. Annu Rev Pathol. 2010;5:99-118. PubMed.
- Ovadya Y, Krizhanovsky V. Senescent cells: SASPected drivers of age-related pathologies. Biogerontology. 2014 Dec;15(6):627-42. Epub 2014 Sep 13 PubMed.
- Zhu Y, Armstrong JL, Tchkonia T, Kirkland JL. Cellular senescence and the senescent secretory phenotype in age-related chronic diseases. Curr Opin Clin Nutr Metab Care. 2014 Jul;17(4):324-8. PubMed.
- Gewirtz DA. Autophagy and senescence: a partnership in search of definition. Autophagy. 2013 May;9(5):808-12. Epub 2013 Feb 19 PubMed.
- Franceschi C, Campisi J. Chronic inflammation (inflammaging) and its potential contribution to age-associated diseases. J Gerontol A Biol Sci Med Sci. 2014 Jun;69 Suppl 1:S4-9. PubMed.
- Giunta B, Fernandez F, Nikolic WV, Obregon D, Rrapo E, Town T, Tan J. Inflammaging as a prodrome to Alzheimer's disease. J Neuroinflammation. 2008;5:51. PubMed.
- Chinta SJ, Lieu CA, Demaria M, Laberge RM, Campisi J, Andersen JK. Environmental stress, ageing and glial cell senescence: a novel mechanistic link to Parkinson's disease?. J Intern Med. 2013 May;273(5):429-36. PubMed.
- Ohta Y, Tremblay C, Schneider JA, Bennett DA, Calon F, Julien JP. Interaction of transactive response DNA binding protein 43 with nuclear factor κB in mild cognitive impairment with episodic memory deficits. Acta Neuropathol Commun. 2014 Apr 1;2:37. PubMed.
Further Reading
Papers
- Chinta SJ, Lieu CA, Demaria M, Laberge RM, Campisi J, Andersen JK. Environmental stress, ageing and glial cell senescence: a novel mechanistic link to Parkinson's disease?. J Intern Med. 2013 May;273(5):429-36. PubMed.
- Tchkonia T, Zhu Y, van Deursen J, Campisi J, Kirkland JL. Cellular senescence and the senescent secretory phenotype: therapeutic opportunities. J Clin Invest. 2013 Mar;123(3):966-72. PubMed.
- Deleidi M, Jäggle M, Rubino G. Immune aging, dysmetabolism, and inflammation in neurological diseases. Front Neurosci. 2015;9:172. Epub 2015 Jun 3 PubMed.
Primary Papers
- Kang C, Xu Q, Martin TD, Li MZ, Demaria M, Aron L, Lu T, Yankner BA, Campisi J, Elledge SJ. The DNA damage response induces inflammation and senescence by inhibiting autophagy of GATA4. Science. 2015 Sep 25;349(6255):aaa5612. PubMed.
- Cassidy LD, Narita M. CELL BIOLOGY. GATA get a hold on senescence. Science. 2015 Sep 25;349(6255):1448-9. PubMed.
Annotate
To make an annotation you must Login or Register.
Comments
This is an interesting paper with lots of data. The concept of NF-κB activation in senescence is relevant to neurodegenerative disorders.
For example, last year we reported that in individuals with mild cognitive impairment there was enhanced NF-κB activation with p65 detection in the nucleus of temporal lobe neurons (Ohta et al., 2014). Moreover, the p65 subunit of NF-κB was detectable in the nucleus of motor neurons from spinal cord samples of human ALS and of mice carrying human transgenes coding for ALS-linked TDP-43 mutants (Swarup et al., 2011, Figs 1 and 2). In these ALS mouse models, the NF-κB activation and neuroinflammation was age-dependent, occurring after 8 months of age when cytoplasmic accumulations of TDP-43 are detected.…More
In light of this paper by Kang et al., it would be of interest to determine if there is a correlation between levels of GATA4 and the appearance of cytoplasmic TDP-43 aggregates, a hallmark of ALS and FTLD.
References:
Ohta Y, Tremblay C, Schneider JA, Bennett DA, Calon F, Julien JP. Interaction of transactive response DNA binding protein 43 with nuclear factor κB in mild cognitive impairment with episodic memory deficits. Acta Neuropathol Commun. 2014 Apr 1;2:37. PubMed.
Swarup V, Phaneuf D, Dupré N, Petri S, Strong M, Kriz J, Julien JP. Deregulation of TDP-43 in amyotrophic lateral sclerosis triggers nuclear factor κB-mediated pathogenic pathways. J Exp Med. 2011 Nov 21;208(12):2429-47. PubMed.
Massachusetts General Hospital
Aging and aging-related neurodegenerative diseases are associated with signs of neuroinflammation. In this new study, Steve Elledge's laboratory in collaboration with Bruce Yankner’s laboratory provide evidence in support of a new molecular mechanism that links the DNA damage response with the senescence-associated secretory phenotype—including expression and secretion of pro-inflammatory cytokines, chemokines and proteases. The authors demonstrate accumulation of the transcription factor GATA4, which is thought to occur through the selective inhibition of autophagy by disrupting GATA4’s association with the autophagy adaptor protein, p62. Moreover, they report increased GATA4 levels in the astrocytes, pyramidal neurons, and oligodendrocytes of aged human frontal cortex in individuals who did not satisfy criteria for a neurodegenerative disease.…More
DNA damage accumulates with aging and is accelerated in brains with Alzheimer’s disease pathology. These exciting discoveries elucidate a new pathway to link DNA damage with neuroinflammation in the brains of presumed clinically normal aged individuals. These findings have important translational potential. Preventing or reversing the accumulation of GATA4 in the aging brain may offer therapeutic benefit by reducing the burden of neuroinflammation and thereby reducing the clinical manifestations of aging and neurodegenerative disease. However, outstanding questions still remain, including what the molecular mechanism of the disruption of the p62 and GATA4 association is, and how to harness this seemingly general mechanism in the appropriate cell type(s) in the nervous system. Also, when will a reliable biomarker for this mechanism be available to characterize the kinetics of this mechanism in the context of the disease spectrum of AD and to design clinical trials with intervention at the optimal stage of the disease?
Make a Comment
To make a comment you must login or register.