Epoxide Hydrolase, a Fat New Target for Alzheimer’s Therapy?
Quick Links
Can making ephemeral anti-inflammatory fatty acids stick around a little longer protect people against Alzheimer’s disease? It just might, according to a study by Hui Zheng and colleagues at the Baylor College of Medicine, Houston, published December 9 in Science Translational Medicine.
- In AD brain and 5xFAD mice, epoxide hydrolase is up and its anti-inflammatory products are down.
- Astrocytes express this lipid metabolism enzyme.
- In mice, inhibiting it restored epoxy fatty acids and a chain of consequences at molecular, synaptic, and behavioral levels of investigation.
These so-called epoxy fatty acids, or EpFAs, protect neurons; alas, epoxide hydrolases quickly break them down. The scientists found more epoxide hydrolase in brain tissue from people with AD and 5xFAD mice than in controls, corresponding to fewer epoxy fatty acids in the mice. Long-term treatment with an inhibitor curbed the hydrolase and boosted EpFA concentration in the mice’s brains. Inhibiting the hydrolase also prevented neuroinflammation and amyloid pathology, synapse damage, and cognitive decline in the mice. Given that epoxide hydrolase inhibitors are already being tested in the clinic for other conditions, they may have the potential to become a new class of AD drug.
Darryl Zeldin, National Institute of Environmental Health Sciences (NIEHS), Research Triangle Park, North Carolina, was unsurprised by these findings. “This is consistent with what we know about the role of inflammation in Alzheimer’s, the effects of epoxide hydrolase inhibitors on epoxide fatty acid levels, and the anti-inflammatory effects of those fatty acids,” he told Alzforum. Aditi Das, University of Illinois at Urbana-Champaign, considers the approach promising. “sEH inhibitors would increase the body’s own bioactive epoxide lipids to a beneficial level, improving our capacity to heal,” she told Alzforum.

Lipid Processing. Arachidonic acid metabolism creates many signaling lipids, including EDP and EET epoxy fatty acids. [Courtesy of Ghosh et al., Science Translational Medicine, 2020.]
Epoxy fatty acids are derived from the omega-6 fatty acid arachidonic acid, which generally is released into the cell in two ways: from endocannabinoids by fatty acid amide hydrolase, or from membrane phospholipids by phospholipase A2. Higher amounts of phospholipase A2 and arachidonic acid have been seen in AD brain tissue and mouse models before (Oct 2008 news). Arachidonic acid metabolism produces various other fatty acids, as well.
Some types of EpFA, such as epoxyeicosatrienoic acids (EETs) and epoxydocosapentaenoic acids (EDPs), are known to be anti-inflammatory and neuroprotective (Node et al., 1999; Wang et al., 2018). Unfortunately, their rapid breakdown by soluble epoxide hydrolase (sEH) limits these beneficial effects. Previously, sEH had been implicated in neurodegenerative diseases because it was found at higher concentrations in people with AD and Parkinson’s disease (Griñán-Ferré et al., 2020; Ren et al., 2018). Inhibiting sEH genetically or with drugs improved the phenotype of mouse models of these diseases, but researchers did not understand how. Zheng wondered if these effects had to do with increased amounts of protective EpFAs.
To find out, first author Anamitra Ghosh and colleagues probed what was happening to sEH and EpFAs in the brains of people with AD and amyloid mouse models. In postmortem AD samples, they confirmed higher expression of PLA2G2A, the gene encoding phospholipase A2, and they also found twice as much sEH as in age-matched controls. Likewise, brain tissue from 4.5-month-old 5xFAD and APPNLGF knock-in mice had more hydrolase and less EET and EDP. Therefore, more hydrolase meant less EpFAs in the brain.
To see which cell types expressed the hydrolase’s gene, Ephx2, the scientists separated astrocytes, microglia, and vascular endothelial cells from 5xFAD and wild-type hippocampus tissue using flow cytometry. Quantitative PCR and immunofluorescence confirmed high Ephx2 expression in astrocytes, as seen before (Lee et al., 2019).
What happens without epoxide hydrolase function? Ghosh and colleagues provoked inflammation in mouse astrocyte cultures with lipopolysaccharide, then treated the cells with the sEH inhibitor TPPU, short for 1-trifluoromethoxylphenyl-3-(1-propionylpiperidin-4-yl) urea. TPPU quelled the inflammation, reducing the cell’s release of nitrite and expression of proinflammatory molecules. Blocking EETs with the antagonist epoxyeicosa-5(Z)-enoic acid abolished the positive effects of TPPU hydrolase inhibition, but adding EETs once again soothed the astrocytes.
TPPU’s anti-inflammatory effects held true in vivo. Feeding wild-type mice LPS provoked inflammation in their cortices and hippocampi; TPPU quelled it. Western blotting and immunofluorescence confirmed both the LPS-induced proinflammatory molecule upregulation and TPPU-induced downregulation. Another sEH inhibitor, called EC5026, showed similar results.
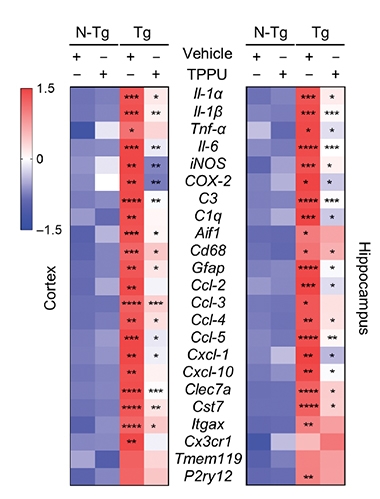
Tamp It Down. In 5xFAD mouse hippocampi, dozens of inflammation markers are upregulated (third column, red) compared to wild-type mice (first and second columns). TPPU turned most of them down (fourth column). [Courtesy of Ghosh et al., Science Translational Medicine, 2020.]
To study long-term effects of epoxide hydrolase inhibition, the scientists spiked the drinking water of 2-month-old 5xFAD mice with TPPU for 2.5 months. They found TPPU in brain tissue by mass spectrometry, indicating it was able to cross the blood-brain barrier. As expected, the treated mice had more EETs and EDPs in their brains.
TPPU treatment changed gene expression in the treated hippocampi. In the 4.5-month-old mice, 73 of the 171 previously upregulated genes had become downregulated. Most of the 73 were in immune-related pathways. Two of them, iNOS and COX-2, were less abundant in the hippocampi of treated mice, as seen via western blotting and immunofluorescence. All told, inhibiting epoxide hydrolase calmed overactive immune pathways in the brains of an amyloidosis mouse model.
The mice’s amyloid plaque burden responded, as well. Ghosh and colleagues stained hippocampal and cortical sections from TPPU-treated and untreated 4.5- and 6.5-month-old 5xFAD mice with the 6E10 plaque antibody. At both time points, treated mice had fewer, smaller plaques and fewer microglia surrounding the plaques than untreated mice, agreeing with previous findings in neuronal cells (Liang et al., 2019). The authors used protein quantification and qPCR analysis to rule out that this was due to off-target effects of TPPU on APP processing or Aβ metabolism.

Astrocytes Agitated, Becalmed. Cyclooxygenase 2 (red), inducible nitric oxide (blue), and GFAP expression were higher in 5xFAD mouse hippocampi (third row) than controls (first and second rows), but TPPU reduced these markers (bottom row). [Courtesy of Ghosh et al., Science Translational Medicine, 2020.]
Frédéric Calon, University of Laval, Quebec, was struck by the effects of TPPU and sEH expression. “I hope this fascinating observation will be replicated in other models,” he told Alzforum.
Next, the scientists stained hippocampal samples from 6.5-month-old 5xFAD mice, either untreated or treated for 4.5 months, with the synaptic marker synaptophysin and the neuronal marker NeuN. They saw that blocking epoxide hydrolase with TPPU partially restored synapse and neuron losses, possibly because treated microglia no longer expressed complement components known to damage synapses, the authors propose.
Did these molecular and cellular changes matter functionally? While untreated 5xFAD mice behaved abnormally on the novel-object and the fear-conditioning tests, TPPU restored their behavior to that of wild-type in both paradigms.
The paper’s overall results are in line with two previous studies that knocked out sEH genetically, not pharmaceutically (Lee et al., 2019; Huang et al., 2018).
Zeldin noted that inhibiting epoxide hydrolase would prevent the breakdown of many additional EpFAs beyond the two in this paper. “The authors did not talk about other EpFAs, such as the omega-3 epoxides, that are prevalent in the brain and would undoubtedly be affected by the sEH inhibitor. These other EpFAs may also be contributing to the phenotypes they report,” he speculated.
Nabil Alkayed, Oregon Health & Science University, Portland, noted that sEH began to climb at 2 months, around the same time astrogliosis and microgliosis start in 5xFAD mice. “It would be important to determine which comes first—whether inflammatory signals induce sEH expression, or sEH upregulation leads to neuroinflammation,” he told Alzforum.
Zeldin wondered how older mice would respond, alluding to potential application in AD patients. “I would like to see what happens if you give an sEH inhibitor to mice that already have inflammation and pathological changes—will inhibiting inflammation then reverse the process?” he asked.
sEH inhibitors have been tried clinically in cardiovascular and renal disorders, though none are FDA-approved; for details, see Alkayed comment below. The new data beg the question of whether sEH may be a plausible target for AD and other brain disorders (Zarriello et al., 2019). “Several sEH inhibitors have produced promising results in Phase 1 studies,” Richard Bazinet, University of Toronto, told Alzforum (full comment below).
One of those studies is led by Bruce Hammock, University of California, Davis, who collaborated with Zheng on this work. His company, EicOsis, is pursuing the sEH inhibitor EC5026 in neuropathic pain (NCT04228302). “In our Phase 1a study, there were no adverse effects reported at doses far above those expected to be therapeutic; however, there are many more steps needed to adequately test safety,” he told Alzforum (full comment below).
One caveat to consider is a known angiogenic effect of inhibiting epoxide hydrolase. Inhibitors are not thought to cause tumors, but they have been reported to stimulate metastasis of existing ones (Panigrahy et al., 2012). “I would be concerned about an older patient with a cold tumor being treated long-term with an sEH inhibitor and having accelerated tumor growth. Screening for cancer becomes really important,” Zeldin explained. Zheng is currently creating analogs of TPPU.—Chelsea Weidman Burke
References
News Citations
Research Models Citations
Antibody Citations
Paper Citations
- Node K, Huo Y, Ruan X, Yang B, Spiecker M, Ley K, Zeldin DC, Liao JK. Anti-inflammatory properties of cytochrome P450 epoxygenase-derived eicosanoids. Science. 1999 Aug 20;285(5431):1276-9. PubMed.
- Wang L, Luo G, Zhang LF, Geng HX. Neuroprotective effects of epoxyeicosatrienoic acids. Prostaglandins Other Lipid Mediat. 2018 Sep;138:9-14. Epub 2018 Jul 18 PubMed.
- Griñán-Ferré C, Codony S, Pujol E, Yang J, Leiva R, Escolano C, Puigoriol-Illamola D, Companys-Alemany J, Corpas R, Sanfeliu C, Pérez B, Loza MI, Brea J, Morisseau C, Hammock BD, Vázquez S, Pallàs M, Galdeano C. Pharmacological Inhibition of Soluble Epoxide Hydrolase as a New Therapy for Alzheimer's Disease. Neurotherapeutics. 2020 Jun 2; PubMed.
- Ren Q, Ma M, Yang J, Nonaka R, Yamaguchi A, Ishikawa KI, Kobayashi K, Murayama S, Hwang SH, Saiki S, Akamatsu W, Hattori N, Hammock BD, Hashimoto K. Soluble epoxide hydrolase plays a key role in the pathogenesis of Parkinson's disease. Proc Natl Acad Sci U S A. 2018 Jun 19;115(25):E5815-E5823. Epub 2018 May 7 PubMed.
- Lee HT, Lee KI, Chen CH, Lee TS. Genetic deletion of soluble epoxide hydrolase delays the progression of Alzheimer's disease. J Neuroinflammation. 2019 Dec 17;16(1):267. PubMed.
- Liang Z, Zhang B, Xu M, Morisseau C, Hwang SH, Hammock BD, Li QX. 1-Trifluoromethoxyphenyl-3-(1-propionylpiperidin-4-yl) Urea, a Selective and Potent Dual Inhibitor of Soluble Epoxide Hydrolase and p38 Kinase Intervenes in Alzheimer's Signaling in Human Nerve Cells. ACS Chem Neurosci. 2019 Sep 18;10(9):4018-4030. Epub 2019 Aug 19 PubMed.
- Huang HJ, Wang YT, Lin HC, Lee YH, Lin AM. Soluble Epoxide Hydrolase Inhibition Attenuates MPTP-Induced Neurotoxicity in the Nigrostriatal Dopaminergic System: Involvement of α-Synuclein Aggregation and ER Stress. Mol Neurobiol. 2018 Jan;55(1):138-144. PubMed.
- Zarriello S, Tuazon JP, Corey S, Schimmel S, Rajani M, Gorsky A, Incontri D, Hammock BD, Borlongan CV. Humble beginnings with big goals: Small molecule soluble epoxide hydrolase inhibitors for treating CNS disorders. Prog Neurobiol. 2019 Jan;172:23-39. Epub 2018 Nov 14 PubMed.
- Panigrahy D, Edin ML, Lee CR, Huang S, Bielenberg DR, Butterfield CE, Barnés CM, Mammoto A, Mammoto T, Luria A, Benny O, Chaponis DM, Dudley AC, Greene ER, Vergilio JA, Pietramaggiori G, Scherer-Pietramaggiori SS, Short SM, Seth M, Lih FB, Tomer KB, Yang J, Schwendener RA, Hammock BD, Falck JR, Manthati VL, Ingber DE, Kaipainen A, D'Amore PA, Kieran MW, Zeldin DC. Epoxyeicosanoids stimulate multiorgan metastasis and tumor dormancy escape in mice. J Clin Invest. 2012 Jan;122(1):178-91. Epub 2011 Dec 19 PubMed.
External Citations
Further Reading
Papers
- Huynh K, Lim WL, Giles C, Jayawardana KS, Salim A, Mellett NA, Smith AA, Olshansky G, Drew BG, Chatterjee P, Martins I, Laws SM, Bush AI, Rowe CC, Villemagne VL, Ames D, Masters CL, Arnold M, Nho K, Saykin AJ, Baillie R, Han X, Kaddurah-Daouk R, Martins RN, Meikle PJ. Concordant peripheral lipidome signatures in two large clinical studies of Alzheimer's disease. Nat Commun. 2020 Nov 10;11(1):5698. PubMed.
Primary Papers
- Ghosh A, Comerota MM, Wan D, Chen F, Propson NE, Hwang SH, Hammock BD, Zheng H. An epoxide hydrolase inhibitor reduces neuroinflammation in a mouse model of Alzheimer's disease. Sci Transl Med. 2020 Dec 9;12(573) PubMed. Correction.
Annotate
To make an annotation you must Login or Register.
Comments
Oregon Health & Science University
The study by Ghosh et al. follows a line of research that started in the early ’90s. It showed that astrocytes are a major source of production of epoxyeicosatrienoates (EETs) in the brain (Davis et al., 2017). EETs are metabolites of arachidonic acid (AA) produced by cytochrome P450 epoxygenase. In acute brain injury such as that which occurs in stroke and traumatic brain injury (TBI), EET’s precursor AA is released from plasma membranes, where it resides as part of membrane phospholipids. AA is subsequently metabolized by multiple enzymes into various eicosanoids that could be detrimental or protective. In chronic neurodegenerative diseases such as Alzheimer's disease and vascular cognitive impairment (VCI), neuroinflammatory signals alter activities of eicosanoid synthetic and metabolizing enzymes, disrupting the balance between neuroprotective and neurotoxic eicosanoids (Zhang et al., 2007).
EETs have been shown to have neuroprotective properties via multiple mechanisms, including vasodilation and anti-inflammation. They have been shown to be protective in stroke (Chen et al., 2020), AD (Liu et al., 2018), and VCI (Pardeshi et al., 2019; Nelson et al., 2014). The levels of EETs are regulated by synthesis and metabolism, and a key enzyme involved in EET’s metabolism is soluble epoxide hydrolase (sEH). In VCI, the expression and activity of sEH have been reported to be increased in cerebrovascular endothelium in postmortem human brain tissue, and genetic polymorphisms have been linked to white-matter hyperintensity (WMH) burden, an early marker and predictor of VCI (Liang et al., 2019).
Inhibition and deletion of sEH have been explored as a therapeutic strategy to increase endogenous EETs bioavailability in multiple brain diseases, especially stroke (Liang et al., 2019). More recently, several studies have demonstrated potential beneficial effects of sEH inhibition in cell-based (Lee et al., 2019) and animal models (Lee et al., 2019; Griñán-Ferré et al., 2020; Tu et al., 2018) of AD.
There have been some clinical trials using at least two different sEH inhibitors: AR9281 was tested in hypertension, but it did not show efficacy (NCT00847899), and GSK2256294 is currently being tested in subarachnoid hemorrhage (NCT03486223) and insulin resistance (NCT03486223). The sEH inhibitor used in the study by Ghosh et al. (TPPU) is not approved for clinical use, but it has been used in experimental models of stroke (Tu et al., 2018), VCI (Griñán-Ferré et al., 2020), and AD (Lee et al., 2019). A third compound, EC5026, also used in the study by Ghosh et al., has completed a Phase Ia safety trial (NCT04228302).
The current study shows that sEH is elevated in postmortem brain tissue from patients with AD and in mouse models of AD, and that long-term sEH inhibition reduces neuroinflammation and Aβ pathology, and improves synaptic integrity and cognitive function in the 5xFAD mouse model of AD. Although confirmatory to previous studies, the study is well-designed and -conducted. Another interesting aspect of the study is its focus on neuroinflammatory mechanisms, showing that astrocytes are the cellular source of sEH and EETs, which then act in autocrine fashion to suppress microglial activation.
Some aspects of the study were not fully explored. For example, in addition to sEH upregulation, the authors found increased expression of PLA2 in postmortem AD human brains, but the mechanisms of upregulation of sEH and PLA2 remain unclear. Since sEH upregulation in this study starts at 2 months, which is around the same time astrogliosis and microgliosis begin in this animal model, it would be important to determine which comes first; i.e., whether inflammatory signals induce sEH expression, or whether sEH upregulation leads to neuroinflammation, and how.
PLA2 upregulation suggests a more generalized alteration in eicosanoid signaling than just EETs/sEH signaling. This is further suggested by the authors’ observation regarding expression of both COX2 and CYP4F8 genes, which produce prostaglandins and hydroxyeicosanoids (such as 20-hydroxyeicosatetraenoate, 20-HETE), respectively. Prostaglandins and 20-HETE are important vasoactive and inflammatory signals, but their role in this study and their potential interactions with EETs/sEH signaling have not been investigated.
Despite the promising results, chronic administration of sEH inhibitors in humans should proceed with caution, especially as a treatment for AD. In a recent study of global cerebral ischemia induced by cardiac arrest in piglets, the same sEH inhibitor TPPU led to neuronal abnormality in sham-treated animals, with neuronal cell body attrition and nuclear condensation. Furthermore, TPPU was ineffective in protecting neurons and reducing neurologic deficit after global cerebral ischemia (O'Brien et al., 2020).
This is reminiscent of another study of cardiac arrest in mice (Hutchens et al., 2008), where sEH knockout mice had greater mortality compared to wild-type mice, raising the possibility that sEH inhibition may interfere with other organ functions (e.g., cardiovascular or pulmonary) required for recovery after cardiac arrest. Although sEH has been extensively investigated as a therapeutic target, its physiological function remains unknown, and therefore, chronic inhibition of sEH may interfere with its function and cause unwanted side effects. Of particular concern in chronic administration of sEH inhibitors is tumorigenesis, given the growth-promoting potential of EETs (Panigrahy et al., 2011; Panigrahy et al., 2012).
References:
Alkayed NJ, Narayanan J, Gebremedhin D, Medhora M, Roman RJ, Harder DR. Molecular characterization of an arachidonic acid epoxygenase in rat brain astrocytes. Stroke. 1996 May;27(5):971-9. PubMed.
Davis CM, Liu X, Alkayed NJ. Cytochrome P450 eicosanoids in cerebrovascular function and disease. Pharmacol Ther. 2017 Nov;179:31-46. Epub 2017 May 18 PubMed.
Zhang W, Koerner IP, Noppens R, Grafe M, Tsai HJ, Morisseau C, Luria A, Hammock BD, Falck JR, Alkayed NJ. Soluble epoxide hydrolase: a novel therapeutic target in stroke. J Cereb Blood Flow Metab. 2007 Dec;27(12):1931-40. Epub 2007 Apr 18 PubMed.
Chen W, Wang M, Zhu M, Xiong W, Qin X, Zhu X. 14,15-Epoxyeicosatrienoic Acid Alleviates Pathology in a Mouse Model of Alzheimer's Disease. J Neurosci. 2020 Oct 14;40(42):8188-8203. Epub 2020 Sep 24 PubMed.
Liu X, Davis CM, Alkayed NJ. P450 Eicosanoids and Reactive Oxygen Species Interplay in Brain Injury and Neuroprotection. Antioxid Redox Signal. 2018 Apr 1;28(10):987-1007. Epub 2017 Apr 20 PubMed.
Pardeshi R, Bolshette N, Gadhave K, Arfeen M, Ahmed S, Jamwal R, Hammock BD, Lahkar M, Goswami SK. Docosahexaenoic Acid Increases the Potency of Soluble Epoxide Hydrolase Inhibitor in Alleviating Streptozotocin-Induced Alzheimer's Disease-Like Complications of Diabetes. Front Pharmacol. 2019;10:288. Epub 2019 Apr 24 PubMed.
Nelson JW, Young JM, Borkar RN, Woltjer RL, Quinn JF, Silbert LC, Grafe MR, Alkayed NJ. Role of soluble epoxide hydrolase in age-related vascular cognitive decline. Prostaglandins Other Lipid Mediat. 2014 Oct;113-115:30-7. Epub 2014 Sep 30 PubMed.
Liang Z, Zhang B, Xu M, Morisseau C, Hwang SH, Hammock BD, Li QX. 1-Trifluoromethoxyphenyl-3-(1-propionylpiperidin-4-yl) Urea, a Selective and Potent Dual Inhibitor of Soluble Epoxide Hydrolase and p38 Kinase Intervenes in Alzheimer's Signaling in Human Nerve Cells. ACS Chem Neurosci. 2019 Sep 18;10(9):4018-4030. Epub 2019 Aug 19 PubMed.
Lee HT, Lee KI, Chen CH, Lee TS. Genetic deletion of soluble epoxide hydrolase delays the progression of Alzheimer's disease. J Neuroinflammation. 2019 Dec 17;16(1):267. PubMed.
Griñán-Ferré C, Codony S, Pujol E, Yang J, Leiva R, Escolano C, Puigoriol-Illamola D, Companys-Alemany J, Corpas R, Sanfeliu C, Pérez B, Loza MI, Brea J, Morisseau C, Hammock BD, Vázquez S, Pallàs M, Galdeano C. Pharmacological Inhibition of Soluble Epoxide Hydrolase as a New Therapy for Alzheimer's Disease. Neurotherapeutics. 2020 Jun 2; PubMed.
Tu R, Armstrong J, Lee KS, Hammock BD, Sapirstein A, Koehler RC. Soluble epoxide hydrolase inhibition decreases reperfusion injury after focal cerebral ischemia. Sci Rep. 2018 Mar 27;8(1):5279. PubMed.
O'Brien CE, Santos PT, Kulikowicz E, Lee JK, Koehler RC, Martin LJ. Neurologic effects of short-term treatment with a soluble epoxide hydrolase inhibitor after cardiac arrest in pediatric swine. BMC Neurosci. 2020 Oct 31;21(1):43. PubMed.
Hutchens MP, Nakano T, Dunlap J, Traystman RJ, Hurn PD, Alkayed NJ. Soluble epoxide hydrolase gene deletion reduces survival after cardiac arrest and cardiopulmonary resuscitation. Resuscitation. 2008 Jan;76(1):89-94. Epub 2007 Aug 28 PubMed.
Panigrahy D, Greene ER, Pozzi A, Wang DW, Zeldin DC. EET signaling in cancer. Cancer Metastasis Rev. 2011 Dec;30(3-4):525-40. PubMed.
Panigrahy D, Edin ML, Lee CR, Huang S, Bielenberg DR, Butterfield CE, Barnés CM, Mammoto A, Mammoto T, Luria A, Benny O, Chaponis DM, Dudley AC, Greene ER, Vergilio JA, Pietramaggiori G, Scherer-Pietramaggiori SS, Short SM, Seth M, Lih FB, Tomer KB, Yang J, Schwendener RA, Hammock BD, Falck JR, Manthati VL, Ingber DE, Kaipainen A, D'Amore PA, Kieran MW, Zeldin DC. Epoxyeicosanoids stimulate multiorgan metastasis and tumor dormancy escape in mice. J Clin Invest. 2012 Jan;122(1):178-91. Epub 2011 Dec 19 PubMed.
University of California
Some sEH inhibitors are in clinical development. Arete Therapeutics took a compound from my laboratory through Phase IIa trials targeting hypertension and diabetes. We learned that this was a poor choice of clinical path and a poor clinical candidate. The sEH inhibitors can work on these targets, but there are competing compounds on the market for these indications that are quite effective. In addition to very rapid metabolism, this compound was reported to not get into the brain.
GSK developed an excellent sEH inhibitor with good pharmacokinetics and efficacy. They targeted COPD, but the compound was dropped for several reasons. Among them was a regulatory decision of wanting to see proof of life span extension—a high bar for a clinical trial. Oregon Health Sciences University has gotten excellent results in a small human clinical trial on stroke with the GSK compound. These data were just announced last month. I would expect that brain penetration would be poor. Possibly if the compound is potent enough it will preserve the epoxy fatty acids in the periphery sufficiently to have a positive CNS effect.
EicOsis Human Health, a company I founded, is developing EC5026 on a clinical path to treat chronic diabetic neuropathy and arthritic pain. One goal is to provide an effective alternative to nonsteroidal and opioid analgesics. EicOsis is developing a related compound for treating arthritis in dogs, pain in cats, and laminitis (neuropathic pain) in horses. As a small company, EicOsis must be very focused on pain.
My academic laboratory, and numerous friends and collaborators around the world, are looking at sEH inhibitors as probes to understand many diseases and as tools to potentially treat diseases. A key action of the compounds is to shift the endoplasmic reticulum stress pathway away from cell senescence and severe inflammation and toward cell survival and resolution of inflammation. Hence the sEH inhibitors act on many chronic diseases ranging from heart failure and fibrosis to diseases of aging and, in particular, those involving chronic neuroinflammation such as Parkinson's, autism, depression, schizophrenia, etc. Certainly neuroinflammation and ER stress contribute to Alzheimer's disease.
Regarding the question of the safety profile of EC5026 and other sEH inhibitors in clinical trials so far, one can never prove a drug safe so we are working quite hard to prove sEH inhibitors to be unsafe. So far we have failed in this. Our studies needed to obtain investigational new drug status from the FDA showed a high safety margin in animals. In human phase 1a single ascending dose studies, there were no adverse effects reported at doses far above those expected to be therapeutic. Although we have FDA "fast track status," there are many more steps needed to adequately test safety in humans.
Regarding inhibiting soluble epoxide hydrolase in Alzheimer’s disease patients, to date, none of our preclinical data suggest downsides to the use of the drug. Worries are, of course, that the drug is stabilizing fundamental biology and resolving inflammation. Inflammation in moderation is a beneficial and critical biological process. The sEH inhibitors appear to resolve deleterious inflammation rather than being anti-inflammatory. Thus we hope that they will be beneficial in diseases involving neuroinflammation. Another concern is that treatment for diseases such as AD will have to be chronic, and some of the patients will be fragile due to age and comorbidities. That said, based on the work from Kenji Hashimoto's laboratory at Chiba University on ER stress in inflammation in the central nervous system, and now the effort of many other investigators, I am optimistic about the value of the sEH inhibitors alone or in combination with other drugs in AD therapy and other chronic diseases associated with aging.
Hashimoto is an expert on chronic CNS diseases; some of his students work on AD. He has shown that sEH inhibitors can reverse and prevent a number of chronic CNS disease in rodent models including autism, Parkinson's, Lewy body disorder, schizophrenia, bipolar, and others. In each case, he showed neuroinflammation and ER stress were critical to the disease. He showed the expected biomarkers, including the sEH protein, went up in inflammation and the natural inflammation-resolving chemical mediators went down in the appropriate brain regions for the disease, and in animals and, in some cases, in human pluripotent stem cells. He also showed that the inflammation-resolving epoxyfatty acids were down in cadaveric human samples of chronic CNS disease. We hope to take the same approach with AD (Hashimoto et al., 2019; Atone et al., 2020).
References:
Hashimoto K. Role of Soluble Epoxide Hydrolase in Metabolism of PUFAs in Psychiatric and Neurological Disorders. Front Pharmacol. 2019;10:36. Epub 2019 Jan 30 PubMed.
Atone J, Wagner K, Hashimoto K, Hammock BD. Cytochrome P450 derived epoxidized fatty acids as a therapeutic tool against neuroinflammatory diseases. Prostaglandins Other Lipid Mediat. 2020 Apr;147:106385. Epub 2019 Nov 5 PubMed.
University of Toronto
Drugs developed for the treatment of Alzheimer’s’ disease have had very limited success, to say the least. This study examining a soluble epoxide hydrolase (sEH) inhibitor is a novel and welcome approach for the field.
The brain is abundant in arachidonic acid, which regulates many functions including inflammation, which, in turn, is thought to contribute to AD. While most famous for its pro-inflammatory products, arachidonic acid can also be converted to epoxy fatty acids. They play important anti-inflammatory roles, keeping balance with the classical products of arachidonic acid.
However, the enzyme sEH rapidly degrades the anti-inflammatory epoxy fatty acids, limiting their ability to counteract inflammation. By adding an sEH inhibitor drug called TTPU orally in mice that have elevated β-amyloid, the team demonstrated that, as expected, epoxy fatty acids are increased in the brain, but also that β-amyloid was reduced as well as markers and neuroinflammation leading to improved synaptic integrity and cognitive behavior scores in the mice.
A surprising and interesting finding is that while the study largely focused on mice, the authors also demonstrated that sEH was elevated in postmortem Alzheimer brains while epoxy fatty acids were lower. Thus, it appears that sEH might be altered in Alzheimer’s and could be a new target for drugs. Clearly, and especially given that other drugs that worked in animal models subsequently failed in human trials, we should not over-extrapolate the current results. However, the study identifies novel testable hypotheses for people with Alzheimer’s.
This is particularly exciting as several sEH inhibitors have produced promising results in Phase 1 human studies. They could be tested in similar animal models to see if they can enter the brain and recapitulate the results observed in the current study with TPPU as we wait for TPPU to enter Phase 1 trials.
Make a Comment
To make a comment you must login or register.