Dysfunctional Lysosomes Cause Ferroptosis in Neurons
Quick Links
A toxic brew of lysosomal lipids, reactive iron atoms, and oxidative stress can spell doom for human neurons. This is the upshot of the first-ever CRISPR screens at the genome-wide level in these cells. Researchers led by Martin Kampmann at the University of California, San Francisco, used the genome-editing tool to dial up or down expression of each protein-coding gene in the human neuronal genome. They uncovered a surprising connection between endolysosomal processing and the iron-dependent cell-death pathway called ferroptosis.
- First genome-wide CRISPR screens in human neurons tweaks gene expression.
- Endolyosomal function, oxidative stress, and iron homeostasis genes key to neuronal survival.
- Sans lysosomal prosaposin, neurons might die by ferroptosis.
- Data available on CRISPRbrain, a communal database.
Zeroing in on that pathway, the researchers found that in the absence of the lysosomal protein prosaposin, glycosphingolipids accumulate in the lysosomes, setting off oxidative stress. This results in a toxic mesh of ferrous ions and peroxidized lipids that can kill neurons. The findings connect pathways that have been implicated separately in neurodegenerative disease, and support the idea that iron-rich “aging pigments,” commonly spotted in older brains, might not be so benign after all.
The findings were posted in a manuscript on bioRxiv on February 5 and the data is the first entered into CRISPRbrain, a communal database in which researchers can share CRISPR screening data gathered from different cell types.
“This is a beautiful study using CRISPRi and CRISPRa to identify genes controlling neuronal response to oxidative stress,” commented Fenghua Hu of Cornell University in Ithaca, New York. “The cell-type-specific effect of gene perturbation is extremely interesting and explains why the neuron is the cell most vulnerable to oxidative stress during neurodegeneration.” CRISPRi and CRISPRa interfere with and activate genes, respectively.
Ellen Sidransky of the National Institutes of Health in Bethesda, Maryland, called the study groundbreaking, noting that it directs attention to the importance of lysosomal pathways in neurodegeneration. “This is an important step toward uncovering molecular mechanisms underlying various neurodegenerative diseases, and the results will likely yield new therapeutic targets,” she wrote.
In its original form, as discovered in bacteria, CRISPR is a way to snip specific regions in the genome with the Cas9 nuclease. Single guide RNAs, affixed with a Cas9 binding sequence, direct the nuclease to specific sequences for cutting. Researchers co-opted the mechanism to edit the genome at will, deleting or replacing nucleic acids of choice. In recent years, researchers have devised multiple variations on this theme, among them CRISPR interference and CRISPR activation. CRISPRi uses a catalytically “dead” form of Cas9 (dCas9) to block transcription of a targeted gene, rather than permanently editing its sequence. In CRISPRa, this dCas9 is fused to an activation domain of a transcription factor, thus revving up expression of targeted genes instead.
By tweaking expression levels, these forms of CRISPR influence the genome similarly to most common genetic variations identified in genome-wide association studies, Kampmann noted. Previously, he and his colleagues had deployed genome-wide CRISPRi and CRISPRa screens in human cancer cell lines (Gilbert et al., 2014). More recently, they used CRISPRi in human induced pluripotent stem cell (iPSC)-derived neurons, targeting a subset of more than 2,000 genes representing the “druggable genome” (Tian et al., 2019).
With this new study, first author Ruilin Tian, now at Southern University of Science and Technology, Shenzhen, China, and colleagues pulled out all the stops. They targeted the entire protein-coding genome in human neurons with CRISPRi and CRISPRa, in search of genes critical to neuron survival.

Screening for Survival. Human iPSCs were transfected to express dCas9 for CRISPRi, or dCas9 fused with a transcription factor for CRISPRa. Later, they were transduced with a library of single guide RNAs, including several for each protein-coding gene in the genome. Fourteen days later, sgRNA frequencies serve as proxies for effects on neuronal survival. [Courtesy of Tian et al., bioRxiv, 2021.]
For both types of screen, the researchers transduced iPSCs with the machinery for CRISPRi or CRISPRa. Later, they transduced the CRISPR-competent iPSCs en masse with a library of sgRNAs targeting each protein-coding gene in the genome. This was done such that only one type of guide RNA infected each cell. They then cultured the cells for two weeks. In that time, cells transduced with sgRNAs targeting essential genes were more likely to die, thus reducing the frequency of that sgRNA in the population of transduced cells. By measuring sgRNAs left on day 14, the researchers could gauge each sgRNA’s influence on survival.
From the CRISPRi screen, genes involved in oxidative stress leapt out as essential for neuronal survival. Superoxide dismutases SOD1 and SOD2 were among the top 10 genes that neurons could not live without. Endolysosomal genes, including subunits of the vacuolar ATPase, which acidifies the lysosome, were also important. Other must-haves included genes involved in cholesterol biosynthesis, iron homeostasis, protein folding, mRNA processing, and autophagy. In the CRISPRa screen, ramping up expression of apoptotic genes docked survival, as expected.
For some genes, including those involved in protein homeostasis, modulating expression in either direction dampened survival, suggesting that neurons narrowly tune their expression. A majority of the genes identified in these screens had not come up in previous CRISPR screens of other cell types, such as cancer cells, suggesting they were uniquely critical for neurons.
Given the prominence of oxidative stress genes that emerged from these survival screens, the researchers ran a series of secondary CRISPR screens to identify genes that influenced survival under conditions of mild oxidative stress, i.e., when the cells were grown in the absence of antioxidants typically added to the media. Under these conditions, they found that Glutathione Peroxidase 4 (GPX4)—an enzyme that uses glutathione to lower the oxidation state of peroxidized lipids—was essential. This enzyme also happens to be essential for putting the kibosh on ferroptosis (Yang et al., 2014).
In fact, oxidized phospholipids are the signature of ferroptosis. In this cell-death pathway, ferrous iron atoms provoke the production of ROS via a chemical process known as the Fenton reaction. This is when iron reacts with hydrogen peroxide formed by incomplete respiration to produce hydroxl radicals. The peroxidized lipids then cause damage to cellular membranes, ultimately rupturing the cells (Dixon et al., 2012).
Focusing on lipid oxidation, Kampmann's group next employed CRISPRi screens to hunt for genes that influenced levels of ROS or peroxidized lipids. Here, they found many expected genes, such as those involved in electron transport or autophagy—pathways that can cause and curtail oxidative stress, respectively. The screens also turned up key regulators of ferroptosis, including GPX4.
One shocker that emerged was prosaposin, a lysosomal protein. In the lysosome, Cathepsin D chops up prosaposin, releasing four activator proteins, saposin A, B, C, D, which assist in the degradation of glycosphingolipids by lysosomal hydrolase enzymes. Without full expression of prosaposin, levels of both ROS and lipid peroxides soared in neurons. Why? Further screens revealed that prosaposin was critical for lysosomal function, but also for keeping reactive iron levels in check inside of neurons.
How was prosaposin involved in all these pathways? To find out, the researchers knocked out the PSAP genes in human iPSCs, and differentiated them into neurons. Compared to their PSAP-replete counterparts, these KO neurons churned out more ROS and had high levels of peroxidized lipids. Interestingly, when the researchers knocked out PSAP from other cell types, including astrocytes and microglia, these toxic signs of stress did not appear, suggesting PSAP specifically kept the peace in neurons.
Bathed in a sea of antioxidants, the PSAP-less neurons survived normally for up to two weeks in culture. However, when the researchers cultured the cells sans antioxidants, they started to die off rapidly after 11 days, and by 14 days, all had perished. Kampmann described the neuronal demise as a messy affair, with neurons appearing to pop.
Apoptosis inhibitors did not save the cells from this fate. Rather, inhibitors of ferroptosis—the iron chelator deferoxamine (DFO) and the lipid peroxidation inhibitor ferrostatin-1—rescued them. This suggested that mild oxidative stress trips off ferroptosis, at least when PSAP is absent.
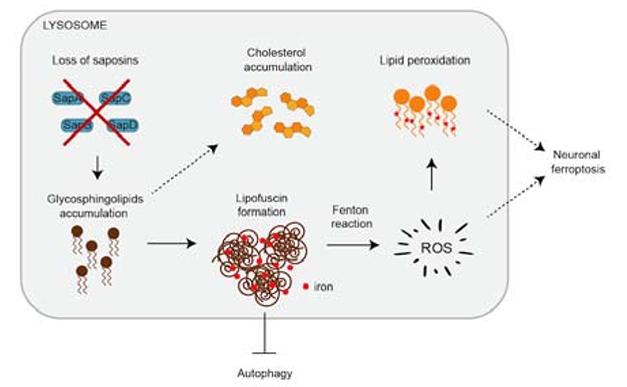
Lyso Lipids Stoke Ferroptosis. Without prosaposin, glycosphingolipids accumulate in the lysosome, which form lipofuscin granules. The iron in these granules produces reactive oxygen species, which oxidize lipids. Peroxidized lipids damage membranes and kill cells via ferroptosis. [Courtesy of Tian et al., bioRxiv, 2021.]

Lipofuscin Pile-Up. Without prosaposin, electron-dense granules of lipids and iron clogged lysosomes of human neurons. [Courtesy of Tian et al., bioRxiv, 2021.]
What is the connection between PSAP and ferroptosis? Examining the PSAP KO neurons via biochemistry, electron microscopy, and super-resolution microscopy, the researchers found that the lysosomes were dramatically enlarged, and chock-full of glycosphingolipids. Strikingly, they found that these lipid-logged organelles were also electron-dense, suggesting they were loaded with iron. In fact, these densities bore an uncanny resemblance to lipid-iron granules called lipofuscin, also known as aging pigment. Lipofuscin soaks up the metal ions from the detritus of iron-rich organelles such as mitochondria, and this iron is thought to provoke the production of ROS via the Fenton reaction.
Strikingly, none of these phenotypes—from lipid accumulation to lysosomal enlargement to lipofuscin or peroxidized lipids—occurred when the researchers knocked PSAP out of other cell types, including iPSC neural progenitor cells, microglia, and astrocytes.
“The unexpected link between saposin-mediated glycosphingolipid degradation, iron metabolism, lipofuscin accumulation, and oxidative stress underscores an important role of lysosomes in regulating iron dynamics in neurons,” wrote Hu.
Could this cascade play out in the aging brain? All of the culprits are there, Kampmann said. For one, oxidative stress is known to rise in the brain with age, and lysosomal function also flags. Levels of not only lipofuscin, but reactive iron increase in aging brains and even more so in neurodegenerative disease (Zhang et al., 2021; Mar 2019 news).
Genetic risk variants for neurodegenerative diseases point to this cascade, as well. Many occur in or around endolysosomal genes. PSAP variants have been linked to lysosomal storage disorders, and, more recently, to Parkinson’s disease (Oji et al., 2020). Sidransky noted that SapC, one of prosaposin’s products, is an essential activator for the enzyme glucocerebrosidase, a lysosomal protein linked to parkinsonism (Jun 2011 news; Jun 2014 news; Oct 2019 news).
Why would these pathways only manifest in neurons? George Perry of the University of Texas, San Antonio, explained that neurons, given their long-lived, post-mitotic nature, have a higher priority to survive than other cells do. As such, they have evolved mechanisms to sequester and detoxify mounting detritus from defunct organelles, and to survive in stressful environs despite faltering function. Perry views lipofuscin as a necessary component of this survival regime. “Lipofuscin has always been relegated to the backwaters of aging research, and this study helps bring it to the forefront,” Perry said. Eventually, the system fails and events such as ferroptosis ensue, he added.
Ashley Bush of the University of Melbourne in Australia made a similar point, noting that despite its dismissal as a meddlesome pigment, lipofuscin is known to concentrate iron. If it was released from these granules in its reactive form, this could explain the inevitable rise in brain iron levels with aging in all mammalian species studied, he wrote (Kurz et al., 2011). The current paper provides evidence for how this could be sinister, rendering neurons vulnerable to ferroptosis under conditions of slight oxidative stress, he added.
The UCSF researchers are continuing to hone their CRISPR tools, and to wield them in different kinds of cells. Case in point: Another manuscript they posted on bioRxiv screened a cell line to look for genes involved in the aggregation of α-synuclein (See et al., 2021). Using a FRET-based sensor, these cells fluoresce when α-synuclein fibrils snap together inside the cell. First author Stephanie See and colleagues uncovered that endolysosomal transport genes had a surprisingly strong sway over α-synuclein aggregation. Notably, they found that a small-molecule inhibitor of PIK-fyve—an endosomal enzyme that mediates endolysosomal trafficking—blocked α-synuclein aggregation. They think it did so by preventing α-synuclein from reaching the lysosome, from where it busts out into the cytoplasm and begins to aggregate.
“Both manuscripts functionally validate strong hits and solidify the endolysosomal network as a hub for neuronal dysfunction in neurodegeneration,” commented Jessica Young of the University of Washington in Seattle.
Interestingly, PIK-fyve inhibitors also reportedly correct autophagy deficits caused by mutations in the C9ORF72 gene. Hexanucleotide repeat expansions in this gene, which cause amyotrophic lateral sclerosis and frontotemporal degeneration, not only derail its endogenous functions of promoting autophagy, but also produce toxic dipeptides that clog the floundering cellular digestive system (Feb 2018 news). In this scenario, PIK-fyve inhibitors promote autophagy.
Separately, PIK-fyve inhibitors have also emerged as a potential treatment for Ebolavirus and SARS-CoV2, presumably by halting the viruses' inexorable march through the endolysosomal system (Kang et al., 2020; clinicaltrials.gov).—Jessica Shugart
References
News Citations
- Does High Iron Push a Person With Pathology Into Dementia?
- Feedback Loop—Molecular Mechanism for PD, Gaucher’s Connection
- Gaucher’s Model Recapitulates Phenotype, Supports Drug Candidate
- Small Molecules Liven Up Lethargic Lysosomes in Parkinson’s Neurons
- Lack of C9ORF72 Protein Renders Neurons More Vulnerable to Degeneration
Paper Citations
- Gilbert LA, Horlbeck MA, Adamson B, Villalta JE, Chen Y, Whitehead EH, Guimaraes C, Panning B, Ploegh HL, Bassik MC, Qi LS, Kampmann M, Weissman JS. Genome-Scale CRISPR-Mediated Control of Gene Repression and Activation. Cell. 2014 Oct 23;159(3):647-61. Epub 2014 Oct 9 PubMed.
- Tian R, Gachechiladze MA, Ludwig CH, Laurie MT, Hong JY, Nathaniel D, Prabhu AV, Fernandopulle MS, Patel R, Abshari M, Ward ME, Kampmann M. CRISPR Interference-Based Platform for Multimodal Genetic Screens in Human iPSC-Derived Neurons. Neuron. 2019 Oct 23;104(2):239-255.e12. Epub 2019 Aug 15 PubMed.
- Yang WS, SriRamaratnam R, Welsch ME, Shimada K, Skouta R, Viswanathan VS, Cheah JH, Clemons PA, Shamji AF, Clish CB, Brown LM, Girotti AW, Cornish VW, Schreiber SL, Stockwell BR. Regulation of ferroptotic cancer cell death by GPX4. Cell. 2014 Jan 16;156(1-2):317-331. PubMed.
- Dixon SJ, Lemberg KM, Lamprecht MR, Skouta R, Zaitsev EM, Gleason CE, Patel DN, Bauer AJ, Cantley AM, Yang WS, Morrison B 3rd, Stockwell BR. Ferroptosis: an iron-dependent form of nonapoptotic cell death. Cell. 2012 May 25;149(5):1060-72. PubMed.
- Zhang G, Zhang Y, Shen Y, Wang Y, Zhao M, Sun L. The Potential Role of Ferroptosis in Alzheimer's Disease. J Alzheimers Dis. 2021;80(3):907-925. PubMed.
- Oji Y, Hatano T, Ueno SI, Funayama M, Ishikawa KI, Okuzumi A, Noda S, Sato S, Satake W, Toda T, Li Y, Hino-Takai T, Kakuta S, Tsunemi T, Yoshino H, Nishioka K, Hattori T, Mizutani Y, Mutoh T, Yokochi F, Ichinose Y, Koh K, Shindo K, Takiyama Y, Hamaguchi T, Yamada M, Farrer MJ, Uchiyama Y, Akamatsu W, Wu YR, Matsuda J, Hattori N. Variants in saposin D domain of prosaposin gene linked to Parkinson's disease. Brain. 2020 Apr 1;143(4):1190-1205. PubMed.
- Kurz T, Eaton JW, Brunk UT. The role of lysosomes in iron metabolism and recycling. Int J Biochem Cell Biol. 2011 Dec;43(12):1686-97. Epub 2011 Sep 3 PubMed.
- See SK, Chen M, Bax S, Tian R, Woerman A, Tse E, Johnson IE, Nowotny C, Muñoz EN, Sengstack J, Southworth DR, Gestwicki JE, Leonetti MD, Kampmann M. PIKfyve inhibition blocks endolysosomal escape of α-synuclein fibrils and spread of α-synuclein aggregation. bioRxiv. January 22, 2021
- Kang YL, Chou YY, Rothlauf PW, Liu Z, Soh TK, Cureton D, Case JB, Chen RE, Diamond MS, Whelan SP, Kirchhausen T. Inhibition of PIKfyve kinase prevents infection by Zaire ebolavirus and SARS-CoV-2. Proc Natl Acad Sci U S A. 2020 Aug 25;117(34):20803-20813. Epub 2020 Aug 6 PubMed.
External Citations
Further Reading
Papers
- Kurz T, Eaton JW, Brunk UT. The role of lysosomes in iron metabolism and recycling. Int J Biochem Cell Biol. 2011 Dec;43(12):1686-97. Epub 2011 Sep 3 PubMed.
- Ashraf A, So PW. Spotlight on Ferroptosis: Iron-Dependent Cell Death in Alzheimer's Disease. Front Aging Neurosci. 2020;12:196. Epub 2020 Jul 14 PubMed.
Primary Papers
- Tian R, Abarientos A, Hong J, Hashemi SH, Yan R, Dräger N, Leng K, Nalls MA, Singleton AB, Xu K, Faghri F, Kampmann M. Genome-wide CRISPRi/a screens in human neurons link lysosomal failure to ferroptosis. Nat Neurosci. 2021 Jul;24(7):1020-1034. Epub 2021 May 24 PubMed.
Annotate
To make an annotation you must Login or Register.
Comments
National Human Genome Research Institute Home
The paper by Tian et al. is truly groundbreaking, because, for the first time, the investigators successfully performed CRISPR-based screens in neurons derived from human induced pluripotent stem cell models, opening many new avenues of research that may lead to an enhanced understanding of the specific functions of human genes in different types of neurons. This is an important step toward uncovering molecular mechanisms underlying various neurodegenerative diseases, and the results will likely yield new therapeutic targets.
Among the strong “hits” were genes influencing reactive oxygen species (ROS), lipid peroxidation, and levels of labile iron. One of these hits, PSAP, encoding the lysosomal protein prosaposin, modifies lysosomal function. Knockdown of PSAP strongly induced ROS, lipid peroxidation, and iron levels, which were aspects not previously associated with this pre-protein.…More
In the lysosome, prosaposin is cleaved by cathepsin into four saposins that are known cofactors for different lysosomal enzymes. In fact, one of the saposins, Sap C, is an essential activator for the enzyme glucocerebrosidase, a known protein linked to parkinsonism. This finding once again directs attention to lysosomal pathways in neurodegeneration.
Overall, the genome-wide CRISPR screen described opens up multiple new paths to pursue, and undoubtedly will lead to many new stories like that of the evolving implications of prosaposin deficiency. The data will be available so that all investigators can interact with this enormous dataset through the new data commons described—CRISPRbrain.
University of Washington
Two new studies from the Kampmann lab use CRISPR technology to uncover regulators of the endolysosomal pathway that impact neurodegeneration. Using CRISPR screens either in hiPSC-derived neurons (Tian et al. 2021) or HEK293 cells (See et al., 2021), they identified modifiers of neurodegenerative phenotypes, including endolysosomal dysfunction, endolysosomal trafficking, and oxidative stress.
This work is exciting because it combines unbiased genome-wide screening with relevant phenotypic readouts implicated in neurodegeneration, moving beyond assays of just cell survival. Both manuscripts functionally validate strong hits and solidify the endolysosomal network as a hub for neuronal dysfunction in neurodegeneration.…More
See et al. implicate inhibition of PIKfyve, a phosphatidylinositol kinase, in mediating a-synuclein aggregation by altering trafficking from early endosomes to lysosomes. Tian et al. show that depletion of PSAP, a lysosomal prosapsosin, increased reactive oxygen species and lysosomal cholesterol, enlarged lysosomes, and increased lipofuscin and iron deposits in neurons.
Many of these phenotypes were not observed non-neuronal cells, including hiPSC-derived microglia, similar to what we have seen for SORL1 depletion in our group (Knupp et al., 2020). This highlights not only the need for cell-type specific assays to understand how disruption of these pathways uniquely affect the diverse cells of the central nervous system, but also to take unique cellular responses into account when designing therapeutic strategies.
Finally, Tian et al., describe CRISPRbrain, a database which focuses on screens done in non-cancerous cells lines. This will be an extremely valuable resource for collating phenotypic data that impact neurodegeneration.
References:
See SK, Chen M, Bax S, Tian R, Woerman A, Tse E, Johnson IE, Nowotny C, Muñoz EN, Sengstack J, Southworth DR, Gestwicki JE, Leonetti MD, Kampmann M. PIKfyve inhibition blocks endolysosomal escape of α-synuclein fibrils and spread of α-synuclein aggregation. bioRxiv. January 22, 2021
Knupp A, Mishra S, Martinez R, Braggin JE, Szabo M, Kinoshita C, Hailey DW, Small SA, Jayadev S, Young JE. Depletion of the AD Risk Gene SORL1 Selectively Impairs Neuronal Endosomal Traffic Independent of Amyloidogenic APP Processing. Cell Rep. 2020 Jun 2;31(9):107719. PubMed.
Rutgers Biomedical & Health Sciences
This is a beautiful study using CRISPRi and CRISPRa to identify genes controlling neuronal response to oxidative stress. The cell type-specific effect of gene perturbation is extremely interesting and explains why the neuron is the most vulnerable cell type to oxidative stress during neurodegeneration.
The unexpected link between saposin-mediated glycosphingolipid degradation, iron metabolism, lipofuscin accumulation, and oxidative stress underscores an important role of lysosome in regulating iron dynamics in neurons.
Finally, CRISPRbrain will be a very useful platform for researchers in the neuroscience field to explore functional genomics screening data.
Northwestern University
The See et al. manuscript builds off of previous work by the Kampmann lab to identify genetic modifiers of neurodegeneration through CRISPR interference (CRISPRi) screens, including the Tian et al. paper. The authors generated a cell line that stably expresses a FRET-based reporter of α-synuclein aggregation, and then, through careful validation experiments, demonstrated that these cells can faithfully report aggregation triggered by the addition of preformed α-synuclein fibrils via a FACS based assay. A genome-wide CRISPRi screen using these cells revealed that phosphatidylinositol kinases are key regulators of α-synuclein aggregation seeded by the extracellularly derived fibrils. Accordingly, both genetic and pharmacologic inhibition of PIKfyve was found to suppress α-synuclein aggregation following addition of recombinant α-synuclein fibrils or fibrils derived from multiple system atrophy (MSA) patient brains.…More
In their discussion, the authors mention that the PIKfyve inhibitor apilimod has been shown to block SARS-CoV-2 infection in vitro and is being evaluated in a COVID19 clinical trial. Recently, a genome-wide CRISPR KO screen to identify SARS-CoV-2 host factors found that several components of the phosphatidylinositol biosynthetic pathway are required for infection with SARS-CoV-2, as well as the coronaviruses HCoV-OC43 and HCoV-229E (Wang et al., 2021). Together these findings raise the intriguing possibility that diverse pathogenic agents that enter cells through the endolysosomal pathway require overlapping mechanisms for exit into the cytosol and propagation to other cells. This suggests that some therapies found to suppress coronavirus infection may hold therapeutic potential for the treatment of synucleinopathies and vice versa. This may be great news for the neurodegeneration field considering the current intense focus on developing antiviral drugs for the treatment of COVID19.
While this study represents a considerable technical achievement, the requirement to use lipofectamine to promote uptake of fibrils into cells limits the ability of the screen to identify genes that regulate α-synuclein uptake. Future studies can overcome this limitation by assessing aggregation in neurons, which should more readily uptake extracellular α-synuclein (Gerdes et al., 2020). As the Tian et al. paper demonstrated the ability to perform genome-wide CRISPRi screens in iPSC-derived neurons, they are well suited to follow up on this exciting work.
References:
Wang R, Simoneau CR, Kulsuptrakul J, Bouhaddou M, Travisano KA, Hayashi JM, Carlson-Stevermer J, Zengel JR, Richards CM, Fozouni P, Oki J, Rodriguez L, Joehnk B, Walcott K, Holden K, Sil A, Carette JE, Krogan NJ, Ott M, Puschnik AS. Genetic Screens Identify Host Factors for SARS-CoV-2 and Common Cold Coronaviruses. Cell. 2021 Jan 7;184(1):106-119.e14. Epub 2020 Dec 9 PubMed.
Gerdes C, Waal N, Offner T, Fornasiero EF, Wender N, Verbarg H, Manzini I, Trenkwalder C, Mollenhauer B, Strohäker T, Zweckstetter M, Becker S, Rizzoli SO, Basmanav FB, Opazo F. A nanobody-based fluorescent reporter reveals human α-synuclein in the cell cytosol. Nat Commun. 2020 Jun 1;11(1):2729. PubMed.
University of Southern California
This elegant screen by Kampmann and colleagues suggests that PIKFYVE inhibition may be able to block the pathological spread of misfolded proteins that cause neurodegeneration (See et al., 2021).
This is an exciting finding because blocking the spread of pathology could greatly modify the course of disease if done early enough. Together with our results showing that PIKFYVE inhibition potently reduces neurodegeneration and pre-existing pathology in C9ORF72 ALS/FTD induced neurons and mice, I think this provides a strong rationale for investigating PIKFYVE inhibition as a therapeutic target for neurodegeneration (Feb 2018 news).
References:
See SK, Chen M, Bax S, Tian R, Woerman A, Tse E, Johnson IE, Nowotny C, Muñoz EN, Sengstack J, Southworth DR, Gestwicki JE, Leonetti MD, Kampmann M. PIKfyve inhibition blocks endolysosomal escape of α-synuclein fibrils and spread of α-synuclein aggregation. bioRxiv. January 22, 2021 …More
The University of Adelaide
This very interesting study adds considerable detail to the emerging picture that lysosomal acidification is intimately linked to cellular iron homeostasis, mitochondrial function, and oxidative stress. As Yambire et al. showed in 2019, reduced lysosomal acidification leads to apparent accumulation of ferric iron in lysosomes and a deficiency of ferrous iron, causing problems in mitochondrial biogenesis and inflammatory responses. (I previously commented on this paper in January 2020).
Tian et al. also describe how correct lysosomal acidification is necessary for cellular iron homeostasis (Weber et al., 2020). It is likely significant that work from Nixon and colleagues has shown that fAD mutations of PSEN1 (Lee et al., 2010) and increased levels of the APP fragment C99 (Jiang et al., 2019)—such as are predicted from many fAD mutations of APP (see Checler et al., 2021)—both reduce lysosomal acidification. So, fAD mutations of PSEN1 and APP may be unified in their effects on iron homeostasis leading to mitochondrial dysfunction, oxidative stress (inducing Aβ production that would also accumulate due to lysosomal dysfunction), and inflammation.…More
The important protective function of Prosaposin in neurons observed by Tian et al. is consistent with results from our earliest transcriptome analyses of knockdown of presenilin 1 or presenilin 2 function in zebrafish embryos using morpholino antisense oligonucleotides. Both treatments caused increased levels of prosaposin transcripts, which can be viewed as a homeostatic response (Newman et al., 2009).
References:
Checler F, Afram E, Pardossi-Piquard R, Lauritzen I. Is γ-secretase a beneficial inactivating enzyme of the toxic APP C-terminal fragment C99?. J Biol Chem. 2021 Jan-Jun;296:100489. Epub 2021 Mar 1 PubMed.
Jiang Y, Sato Y, Im E, Berg M, Bordi M, Darji S, Kumar A, Mohan PS, Bandyopadhyay U, Diaz A, Cuervo AM, Nixon RA. Lysosomal Dysfunction in Down Syndrome Is APP-Dependent and Mediated by APP-βCTF (C99). J Neurosci. 2019 Jul 3;39(27):5255-5268. Epub 2019 May 1 PubMed.
Lee JH, Yu WH, Kumar A, Lee S, Mohan PS, Peterhoff CM, Wolfe DM, Martinez-Vicente M, Massey AC, Sovak G, Uchiyama Y, Westaway D, Cuervo AM, Nixon RA. Lysosomal proteolysis and autophagy require presenilin 1 and are disrupted by Alzheimer-related PS1 mutations. Cell. 2010 Jun 25;141(7):1146-58. PubMed.
Newman M, Tucker B, Nornes S, Ward A, Lardelli M. Altering presenilin gene activity in zebrafish embryos causes changes in expression of genes with potential involvement in Alzheimer's disease pathogenesis. J Alzheimers Dis. 2009;16(1):133-47. PubMed.
Weber RA, Yen FS, Nicholson SP, Alwaseem H, Bayraktar EC, Alam M, Timson RC, La K, Abu-Remaileh M, Molina H, Birsoy K. Maintaining Iron Homeostasis Is the Key Role of Lysosomal Acidity for Cell Proliferation. Mol Cell. 2020 Feb 6;77(3):645-655.e7. Epub 2020 Jan 23 PubMed.
Yambire KF, Rostosky C, Watanabe T, Pacheu-Grau D, Torres-Odio S, Sanchez-Guerrero A, Senderovich O, Meyron-Holtz EG, Milosevic I, Frahm J, West AP, Raimundo N. Impaired lysosomal acidification triggers iron deficiency and inflammation in vivo. Elife. 2019 Dec 3;8 PubMed.
Make a Comment
To make a comment you must login or register.