Does LRRK2 Sweep α-Synuclein from the Cell?
Quick Links
Mutations in the LRRK2 gene spike the risk of Parkinson’s disease, but researchers still do not understand how. Two recent studies strengthen the idea that LRRK2 modulates the endolysosomal disposal system of the cell. In the September 12 Proceedings of the National Academy of Sciences, researchers led by Takeshi Iwatsubo at the University of Tokyo reported that protein accumulation in lysosomes leads LRRK2 to swoop in and prod these organelles to disgorge their contents into the extracellular space. In theory, this jetsam could be taken up by other cells, helping to propagate misfolded proteins, including, but not limited to, α-synuclein, Iwatsubo suggested.
- LRRK2 encourages stressed lysosomes to dump their garbage outside cells.
- It also promotes secretion of α-synuclein from endosomes.
- The findings strengthen the case for treating PD with LRRK2 inhibitors.
A second study provides direct evidence for this. Researchers led by Seung-Jae Lee at Seoul National University College of Medicine, South Korea, report that LRRK2 diverts α-synuclein from lysosomal degradation and toward exocytosis. LRRK2 promoted the propagation of toxic α-synuclein in rodent, worm, and cellular models, and PD variants of the kinase supercharged this process, according to their August 27 Nature Communications paper.
Scientists said these findings support the development of LRRK2 inhibitors underway at several pharmaceutical companies. “LRRK2 inhibitors could potentially block α-synuclein spreading, not just for LRRK2 mutation-associated disease, but also synucleinopathies in general,” said Zhenyu Yue at the Icahn School of Medicine at Mount Sinai in New York.
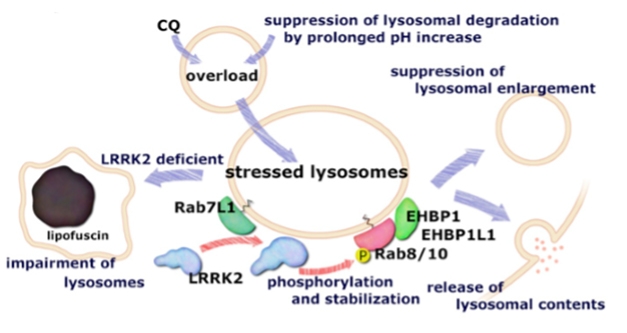
Stress Relief. LRRK2 and Rab8/10 stimulate overloaded lysosomes to spit their contents from cells, returning them to normal size and function. [Courtesy of Eguchi et al., PNAS.]
Several previous studies connected LRRK2 to lysosomal dysfunction, but details were lacking (Jun 2012 news; Mar 2013 conference news). To get a better idea of what LRRK2 does at the lysosome, Iwatsubo and colleagues used mouse fibroblasts, macrophages, and microglia, cell types that all produce large amounts of endogenous LRRK2, as well as HEK293 cells that overexpressed transgenic LRRK2. Joint first authors Tomoya Eguchi, Tomoki Kuwahara, and Maria Sakurai stressed the cells with the antimalarial drug chloroquine. At acidic pH, this small molecule becomes protonated and less lipid-soluble. It builds up in lysosomes, enlarging them, stopping them from degrading debris, and prompting them to fuse with the cell membrane, whereupon they spill their contents. Lysosomal exocytosis also occurs in normal physiology, and aids processes such as membrane repair and myelination (Reddy et al., 2001; Samie and Xu, 2014).
Eguchi and colleagues found that LRRK2 docked to the outer surface of lysosomes within a few hours after exposure to chloroquine. Other chemicals that stress cells but do not swell these organelles, such as lysosomal inhibitors or hydrogen peroxide, did not elicit this response, whereas the yeast polysaccharide zymosan, another agent that engorges the acidic vesicles, did. LRRK2 seems to be selectively recruited to overstuffed lysosomes, the authors concluded.
What does LRRK2 do there? When the authors knocked down the protein, the organelles grew even more as they stopped disgorging their contents to the extracellular space. On the other hand, PD variants of LRRK2 that have heightened kinase activity kept lysosome size in check. These variants also enhanced lysosomal exocytosis. The results suggested that LRRK2 relieves lysosome overload by stimulating them to jettison their contents from the cell.
The authors examined proteins known to interact with LRRK2 to decipher the mechanism. They found that the trafficking protein Rab29, aka Rab7L1, moved first to swollen lysosomes, recruiting LRRK2. Downstream, LRRK2 phosphorylated the GTPases Rab8 and Rab10, attracting them to lysosomes as well, and holding them there. The authors knocked down each of these Rabs in turn and found that Rab8 activity kept lysosomes small, whereas Rab10 promoted their extracellular secretion. To see whether something similar happened in vivo, the authors injected young mice with chloroquine. Kidney cells in LRRK2 knockouts accumulated more waste than did those cells in heterozygous littermates, in keeping with the idea that LRRK2 helps relieve lysosomal stress.
“Chronic upregulation of [lysosomal exocytosis] during neurodegenerative disease may reduce degradation within neuronal lysosomes, and increase release of poorly digested synuclein aggregates into the extracellular space,” Iwatsubo wrote to Alzforum. In addition to encouraging propagation of misfolded proteins, this could release lysosomal proteases that might stimulate inflammation, he suggested. He plans to measure whether LRRK2 stimulates α-synuclein release in stressed cells.
Others agreed that LRRK2 might help orchestrate the cellular response to stress. However, they noted that chloroquine is artificial, was used at very high concentration, and has pleiotropic effects. “It will be important to determine if more physiological stimuli induce similar lysosomal overload,” Sabine Hilfiker at the Institute of Parasitology and Biomedicine, Spanish National Research Council, Granada, wrote to Alzforum (see comment below).

Halting Propagation.
In cell cultures containing pathogenic G2019S LRRK2 (top), α-synuclein aggregates (green) propagate to new cells (nuclei are blue). Inhibiting LRRK2 (bottom) stops this transfer. [Courtesy of Bae et al., Nature Communications.]
Lee and colleagues focused on propagation of misfolded α-synuclein. First authors Eun-Jin Bae and Dong-Kyu Kim injected a virus expressing human α-synuclein into the vagus nerves in the necks of wild-type and LRRK2 knockout rats. In this propagation model, the vagus nerve transports the virus to the medulla oblongata, resulting in selective expression of α-synuclein in medullary neurons. From there, α-synuclein aggregates gradually spread to more rostral brain regions (Ulusoy et al., 2013). However, Bae and Kim found that eight and 12 weeks after injection, knockouts sported fewer axons containing α-synuclein in the pons, midbrain, and forebrain than did wild-type mice. These LRRK2 findings held across species. Worms tend to accumulate α-synuclein deposits with age, but those lacking the LRRK2 analogue LRK-1 developed fewer.
How might LRRK2 promote spreading? Previous work suggested that aggregated α-synuclein passes between cells via exocytosis and endocytosis (for review see Lee et al., 2014). To examine this, the authors turned to SH-SY5Y neuronal cell lines engineered to overexpress human LRRK2 variants. The authors expressed labeled α-synuclein in these and wild-type SH-SY5Y cells, which make little LRRK2. Cells overexpressing the kinase secreted more α-synuclein into the media than did controls. LRRK2 cells also took up more α-synuclein. The authors measured this by fluorescence complementation, whereby α-synuclein in donor cells was labeled with one half of a fluorophore, while that in recipient cells was labeled with the other half, and only when the two merged did they fluoresce. LRRK2-expressing recipient cells lit up with numerous bright spots, whereas controls had almost none. This supports the idea that LRRK2 stimulates α-synuclein spreading between cells. Cells expressing the PD LRRK2 variant, G2019S, secreted and took up about 50 percent more aggregated α-synuclein than did cells expressing wild-type LRRK2. Inhibiting the kinase, on the other hand, blocked secretion (see image above).
Like Iwatsubo and colleagues, Lee and colleagues found that LRRK2 acted through endosomal Rabs. However, Rab35 appeared to be the main effector. It turned up in endosomes along with α-synuclein and LRRK2. A dominant negative Rab35 suppressed α-synuclein propagation in SH-SY5Y cells. In worms lacking Rab35, little α-synuclein accumulated with age, while LRK-1 knockouts transfected with constitutively active Rab35 amassed α-synuclein inclusions.
Finally, the authors tested LRRK2 inhibition in vivo. They used mice that express human α-synuclein under a Thy1 promoter and develop large endosomes stuffed with α-synuclein and decorated with Rab35 (Rockenstein et al., 2002). The authors injected an LRRK2 inhibitor into the mice for four weeks, and found it suppressed Rab35 to wild-type levels and nearly abolished endosomal α-synuclein. Instead, α-synuclein appeared in lysosomes, suggesting it was being degraded. Treated mice had fewer α-synuclein inclusions than untreated, although still more than wild-type controls.
“Our data suggest that LRRK2 functions as a checkpoint regulator in the endolysosomal pathway. It determines whether cargo should be degraded, or take an alternative pathway,” Lee wrote to Alzforum.
Dario Alessi at the University of Dundee, Scotland, U.K., found the data intriguing. “This study is quite elegant in that they use two animal models and come up with the same answer, that phosphorylation of Rab35 is critical for spreading α-synuclein,” he told Alzforum. His group collaborates with the Michael J. Fox Foundation and Abcam to develop antibodies to various LRRK2-phosphorylated Rabs. “Rab35 hadn’t been prioritized, but based on these results, we’ve initiated a new program to make Rab35 antibodies,” he said. This reagent will help researchers study the role of the protein in vivo, which will be critical in assessing its effects.
“It’s hard to know how these data will translate to people,” Alessi said, noting that the effects of LRRK2 and Rab35 on α-synuclein propagation in animal models were modest. Other researchers proposed repeating the cell-culture findings from both papers in human iPSC-derived neurons.
While LRRK2 mutations occur in only 2 to 4 percent of PD cases (Feb 2012 news), another recent paper hints that these findings might be relevant for sporadic PD. Analyzing postmortem brains of people with idiopathic PD, researchers led by Timothy Greenamyre at the University of Pittsburgh found an increase in LRRK2 kinase activity in dopaminergic neurons in the substantia nigra. Alessi was a co-author. First author Roberto Di Maio and colleagues saw a similar activation in rats treated with the dopaminergic neuron toxin, rotenone, and in rats that overexpress synuclein. The uptick in LRRK2 activity correlated with lysosomal abnormalities and a boost in phosphorylated Rab10 (Di Maio et al., 2018). LRRK2 variants also associate with other disorders, such as Crohn’s disease and multiple system atrophy (Jan 2018 news; Heckman et al., 2014).
Denali Therapeutics has a LRRK2 inhibitor, DNL201, which recently met its endpoints in a Phase 1a study, and will advance to Phase 1b testing in PD patients (Aug 1 press release). Several other pharma companies recently presented preclinical data on LRRK2 inhibitors at the Biennial International LRRK2 meeting, held September 2–4 in Padua, Italy. “By next year, it wouldn’t surprise me to see half a dozen companies doing trials with inhibitors,” Alessi said. “There’s huge excitement in the field about their potential.”
That excitement may have been tempered by a recent report linking LRRK2 knockout to lysosomal dysfunction and age-dependent dopaminergic neurodegeneration in mice (Giaime et al., 2017). However, a massive genetics study led by Mark Cookson at the NIA, Bethesda, Maryland, which comprised three different PD genetics consortia, concluded that LRRK2 loss of function is unlikely to cause Parkinson’s. Comparing more than 11,000 cases and more than 12,000 controls, joint first authors Cornelis Blauwendraat and Xylena Reed found no association between LRRK2 or LRRK1 loss-of-function variants and PD, even though cell lines from several carriers suggested protein levels were reduced by half. The researchers concluded that kinase inhibitor or allele-specific targeting of mutant LRRK2 remain viable therapeutic strategies for PD.—Madolyn Bowman Rogers and Tom Fagan
References
News Citations
- Evidence Piles Up for Lysosomal Dysfunction in Parkinson’s
- LRRK Watchers’ Eyes Turn to Inflammation, Autophagy, Kinase
- More Than a LRRK: PD Field Thinks Big With Genetic Cohort
- Genetic Findings Link Parkinson’s to Crohn’s Disease, Inflammation
Paper Citations
- Reddy A, Caler EV, Andrews NW. Plasma membrane repair is mediated by Ca(2+)-regulated exocytosis of lysosomes. Cell. 2001 Jul 27;106(2):157-69. PubMed.
- Samie MA, Xu H. Lysosomal exocytosis and lipid storage disorders. J Lipid Res. 2014 Jun;55(6):995-1009. Epub 2014 Mar 25 PubMed.
- Ulusoy A, Rusconi R, Pérez-Revuelta BI, Musgrove RE, Helwig M, Winzen-Reichert B, Monte DA. Caudo-rostral brain spreading of α-synuclein through vagal connections. EMBO Mol Med. 2013 Jul;5(7):1119-27. PubMed.
- Lee HJ, Bae EJ, Lee SJ. Extracellular α--synuclein-a novel and crucial factor in Lewy body diseases. Nat Rev Neurol. 2014 Feb;10(2):92-8. Epub 2014 Jan 28 PubMed.
- Rockenstein E, Mallory M, Hashimoto M, Song D, Shults CW, Lang I, Masliah E. Differential neuropathological alterations in transgenic mice expressing alpha-synuclein from the platelet-derived growth factor and Thy-1 promoters. J Neurosci Res. 2002 Jun 1;68(5):568-78. PubMed.
- Di Maio R, Hoffman EK, Rocha EM, Keeney MT, Sanders LH, De Miranda BR, Zharikov A, Van Laar A, Stepan AF, Lanz TA, Kofler JK, Burton EA, Alessi DR, Hastings TG, Greenamyre JT. LRRK2 activation in idiopathic Parkinson's disease. Sci Transl Med. 2018 Jul 25;10(451) PubMed.
- Heckman MG, Schottlaender L, Soto-Ortolaza AI, Diehl NN, Rayaprolu S, Ogaki K, Fujioka S, Murray ME, Cheshire WP, Uitti RJ, Wszolek ZK, Farrer MJ, Sailer A, Singleton AB, Chinnery PF, Keogh MJ, Gentleman SM, Holton JL, Aoife K, Mann DM, Al-Sarraj S, Troakes C, Dickson DW, Houlden H, Ross OA. LRRK2 exonic variants and risk of multiple system atrophy. Neurology. 2014 Dec 9;83(24):2256-61. Epub 2014 Nov 5 PubMed.
- Giaime E, Tong Y, Wagner LK, Yuan Y, Huang G, Shen J. Age-Dependent Dopaminergic Neurodegeneration and Impairment of the Autophagy-Lysosomal Pathway in LRRK-Deficient Mice. Neuron. 2017 Nov 15;96(4):796-807.e6. Epub 2017 Oct 19 PubMed.
External Citations
Further Reading
Primary Papers
- Eguchi T, Kuwahara T, Sakurai M, Komori T, Fujimoto T, Ito G, Yoshimura SI, Harada A, Fukuda M, Koike M, Iwatsubo T. LRRK2 and its substrate Rab GTPases are sequentially targeted onto stressed lysosomes and maintain their homeostasis. Proc Natl Acad Sci U S A. 2018 Sep 12; PubMed.
- Bae EJ, Kim DK, Kim C, Mante M, Adame A, Rockenstein E, Ulusoy A, Klinkenberg M, Jeong GR, Bae JR, Lee C, Lee HJ, Lee BD, Di Monte DA, Masliah E, Lee SJ. LRRK2 kinase regulates α-synuclein propagation via RAB35 phosphorylation. Nat Commun. 2018 Aug 27;9(1):3465. PubMed.
- Di Maio R, Hoffman EK, Rocha EM, Keeney MT, Sanders LH, De Miranda BR, Zharikov A, Van Laar A, Stepan AF, Lanz TA, Kofler JK, Burton EA, Alessi DR, Hastings TG, Greenamyre JT. LRRK2 activation in idiopathic Parkinson's disease. Sci Transl Med. 2018 Jul 25;10(451) PubMed.
- Blauwendraat C, Reed X, Kia DA, Gan-Or Z, Lesage S, Pihlstrøm L, Guerreiro R, Gibbs JR, Sabir M, Ahmed S, Ding J, Alcalay RN, Hassin-Baer S, Pittman AM, Brooks J, Edsall C, Hernandez DG, Chung SJ, Goldwurm S, Toft M, Schulte C, Bras J, Wood NW, Brice A, Morris HR, Scholz SW, Nalls MA, Singleton AB, Cookson MR, COURAGE-PD (Comprehensive Unbiased Risk Factor Assessment for Genetics and Environment in Parkinson’s Disease) Consortium, the French Parkinson’s Disease Consortium, and the International Parkinson’s Disease Genomics Consortium (IPDGC). Frequency of Loss of Function Variants in LRRK2 in Parkinson Disease. JAMA Neurol. 2018 Nov 1;75(11):1416-1422. PubMed.
Annotate
To make an annotation you must Login or Register.
Comments
IPBLN, CSIC (Spanish National Research Council)
Two recent studies report on the role of LRRK2 in altering endolysosomal structure/function and lysosomal secretion, albeit coming to different conclusions. Eguchi et al. used various tissue culture cell lines treated with chloroquine, a lysosomotropic agent that enlarges lysosomal structures and causes lysosomal overload stress, as measured by the secretion of cathepsin D. Their data indicate that knockdown of endogenous LRRK2 or inhibition of the LRRK2 kinase activity by pharmacological means causes a further increase in the size of lysosomal structures in the presence of chloroquine, associated with a decrease in the secretion of cathepsin D. They further report that overexpression of pathogenic LRRK2 mutants rescues the chloroquine-induced lysosomal structural alterations, even though the effect of these mutants on cathepsin D secretion was not addressed. The authors further link these effects to a Rab7L1-mediated recruitment of LRRK2 to the chloroquine-enlarged lysosomes, as well as to the involvement of Rab8/Rab10, two protein substrates for the kinase activity of LRRK2. The latter link requires further validation, since knockdown of Rab8 was found to cause lysosomal enlargement without altered cathepsin D secretion, whilst knockdown of Rab10 caused no lysosomal enlargement but had an effect on cathepsin D secretion, suggesting that these two readouts may not go hand in hand. In addition, the nature of the enlarged structures needs further clarification using additional lysosomal markers apart from LAMP1. This is especially important since the chloroquine-mediated release of intermediates of cathepsin D processing suggests that the released species may reside in compartments other than lysosomes. Whilst it will be important to determine whether the same findings can be observed with other, more physiological stimuli to induce lysosomal overload, these data suggest that inhibiting LRRK2 kinase impairs lysosomal secretion under lysosomal overload stress conditions.
Similarly, Bae et al., using different model systems and technical approaches, showed that knockout of LRRK2 in either C. elegans or rat models impairs α-synuclein propagation, which involves exocytosis of α-synuclein from donor cells and subsequent endocytosis by recipient cells. Importantly, in cultured cells, their data further indicate that pathogenic LRRK2 increases uptake in recipient cells as well as release from donor cells. These studies were performed using a bimolecular fluorescence complementation assay, which, due to the irreversible nature of the interactions, may overestimate the observed effects. In addition, the α-synuclein species which is transmitted from one cell to another in those assays remains unclear. Nevertheless, an LRRK2 kinase inhibitor was found to reverse the effects of pathogenic LRRK2 on α-synuclein propagation in vitro, and importantly also decreased the α-synuclein aggregates in the brains of α-synuclein transgenic mice. The authors further show that the effects on α-synuclein propagation in worms and in cell culture models are modulated when altering the levels and/or activity of Rab35, another reported protein substrate for the LRRK2 kinase activity. Their data altogether suggest that pathogenic LRRK2 causes increased α-synuclein propagation, presumably via increased lysosomal exocytosis, even though this is not formally demonstrated.
The two studies differ in the set of LRRK2 Rab protein substrates that they nominate as regulators for the trafficking events analyzed. It remains to be seen whether this is due to studying distinct intracellular compartments (e.g., lysosomes versus phagolysosomes versus late endosomes) and/or distinct cell types. In addition, caution should be exercised when expressing phosphomimetic or phosphodeficient Rab variants, as such mutations can cause loss-of-function protein variants in all cases. Therefore, formal proof that the observed intracellular trafficking deficits are mediated by LRRK2-mediated phosphorylation of these distinct Rab proteins is largely lacking. Whilst both reports clearly nominate LRRK2 as a regulator of proper lysosomal functioning, additional cellular studies performed under endogenous levels of pathogenic LRRK2 expression and measuring endogenous α-synuclein accumulation and secretion, as recently reported (Schapansky et al., 2018), combined with a careful analysis of Rab protein substrate phosphorylation and localization using appropriate tools such as phospho-state-specific antibodies, may shed conclusive insights into the mechanism(s) by which pathogenic LRRK2 regulates endolysosomal homeostasis and α-synuclein propagation.
References:
Schapansky J, Khasnavis S, DeAndrade MP, Nardozzi JD, Falkson SR, Boyd JD, Sanderson JB, Bartels T, Melrose HL, LaVoie MJ. Familial knockin mutation of LRRK2 causes lysosomal dysfunction and accumulation of endogenous insoluble α-synuclein in neurons. Neurobiol Dis. 2018 Mar;111:26-35. Epub 2017 Dec 12 PubMed.
Indiana University School of Medicine
Both Bae et al. and Eguchi et al. show LRRK2 functioning in the endocytic recycling pathway, which is consistent with an earlier study by Mark Cookson’s group showing that LRRK2 interacts with Rab7L1—a protein involved in endosomal recycling (Beilina et al., 2014). A number of other subsequent studies reported similar findings, albeit some studies located LRRK2 functioning at different steps in the endocytic recycling pathway.
These subtle differences are also apparent in these two papers, where Bae et al. show LRRK2 functioning at the endosome in concert with Rab35, while Eguchi et al. show LRRK2 interacting with stressed lysosome via Rab7L1. However, both papers show that LRRK2 kinase activity enhances exocytosis—although of different organelles. It is not immediately clear what caused the difference in LRRK2 localization between these studies; however, since they used different cells lines and studied under different conditions—one used chloroquine to induce lysosomal stress and the other used exogenous α-synuclein aggregates to track propagation—they might have been observing different LRRK2 conformations (potentially a GTP-bound versus a GDP-bound conformation).
References:
Beilina A, Rudenko IN, Kaganovich A, Civiero L, Chau H, Kalia SK, Kalia LV, Lobbestael E, Chia R, Ndukwe K, Ding J, Nalls MA, International Parkinson’s Disease Genomics Consortium, North American Brain Expression Consortium, Olszewski M, Hauser DN, Kumaran R, Lozano AM, Baekelandt V, Greene LE, Taymans JM, Greggio E, Cookson MR. Unbiased screen for interactors of leucine-rich repeat kinase 2 supports a common pathway for sporadic and familial Parkinson disease. Proc Natl Acad Sci U S A. 2014 Feb 18;111(7):2626-31. Epub 2014 Feb 7 PubMed.
Evotec
We think that the papers are very nice! Eguchi et al. confirm all our recently published findings and they observe an increase in Rab10 phosphorylation upon lysosomal activation, which is very intriguing. This is the very first time that we have seen a stimulus of endogenous LRRK2 activity (recent work from Dario Alessi's group shows that Rab29 overexpression stimulates LRRK2).
They also find that the EHBPL1 protein is important for their lysosomal phenotype and, interestingly, we identified this protein as phospho-specific interactor (Steger et al., 2017). Bae et al. show that increased LRRK2 activity correlates with increased α-syn deposition and that this is related to defects in the lysosomal machinery (Rab35-mediated). It would be nice to test whether Rabs other than Rab35, especially Rab3a (strongly expressed in the brain), as well as Rab10 and Rab29, are important regulators of this phenotype. Also, it would be important to show that Rab35 is directly phosphorylated by LRRK2 in this system by using a global/targeted phosphoproteomics approach. There are also phospho-specific Rab antibodies available that could be used to partially answer this question (Lis et al., 2018).
Overall, we don't think that the conclusions of the two papers are very different. Yes, they use different models, but they both show that LRRK2 is involved in the lysosomal pathway and that pathogenic LRRK2 promotes the secretion of lysosomal contents (hence kinase activity-dependent). They are very nice follow ups of our published work (Steger et al., 2017; Steger et al., 2016).
We think that one should analyze the Rab-LRRK2 pathway in more detail in more tissues and cell types. For example, why are dopaminergic neurons of the substantia nigra preferentially affected by Parkinson's disease? There might be specific Rab subtypes (and effectors) that are responsible for this. Moreover, we know that LRRK2 is expressed at high levels in tissues other than brain. We think that LRRK2 function in these cell types (e.g., immune cells, intestine, and lungs) might be important for PD pathogenesis and that proteomic and cellular approaches are needed to answer these open questions. Finally, because of these recent studies, we think that LRRK2 might be linked to PARKIN-PINK1 mediated mitophagy.
— Mathias Mann is the co-author of this comment.
References:
Steger M, Diez F, Dhekne HS, Lis P, Nirujogi RS, Karayel O, Tonelli F, Martinez TN, Lorentzen E, Pfeffer SR, Alessi DR, Mann M. Systematic proteomic analysis of LRRK2-mediated Rab GTPase phosphorylation establishes a connection to ciliogenesis. Elife. 2017 Nov 10;6 PubMed.
Lis P, Burel S, Steger M, Mann M, Brown F, Diez F, Tonelli F, Holton JL, Ho PW, Ho SL, Chou MY, Polinski NK, Martinez TN, Davies P, Alessi DR. Development of phospho-specific Rab protein antibodies to monitor in vivo activity of the LRRK2 Parkinson's disease kinase. Biochem J. 2018 Jan 2;475(1):1-22. PubMed.
Steger M, Tonelli F, Ito G, Davies P, Trost M, Vetter M, Wachter S, Lorentzen E, Duddy G, Wilson S, Baptista MA, Fiske BK, Fell MJ, Morrow JA, Reith AD, Alessi DR, Mann M. Phosphoproteomics reveals that Parkinson's disease kinase LRRK2 regulates a subset of Rab GTPases. Elife. 2016 Jan 29;5 PubMed.
Make a Comment
To make a comment you must login or register.