Does Amyloid Shrink Neurons, Shorten Their Fuses?
Quick Links
Neurons can be quick to fire while in their death throes. This may be because their reduced size gives them a shorter fuse, according to a study published November 13 in Neuron. Researchers led by Stefan Remy of the German Center for Neurodegenerative Diseases in Bonn reported that as amyloid plaques overrun the brain in a mouse model of Alzheimer’s disease, shrinking dendrites render hippocampal neurons small enough that they are more easily stimulated, firing more often. Both dendritic destruction and hyperactivity have been observed in the context of neurodegenerative diseases, including AD, but this study proposes a mechanistic link between the two processes.
“Finding this structure-function relation is very exciting and represents a major advance in our understanding of how amyloid-β pathology alters brain function at the level of individual neurons,” Marc Busche of the Technical University of Munich wrote in a comment to Alzforum. “This novel mechanism may also have broad implications for other diseases that are associated with neurodegeneration.” Busche was not an author on the paper.

Burst Boost. Local field potentials measured in the hippocampus revealed network bursts in APP/PS1 mice (right) compared with the single action potentials seen in wild-type mice (left). [Image courtesy of Zuzana Šišková et al., Neuron, 2014.]
Long before neurons croak in the face of disease, they start to shrink and change shape, Remy told Alzforum. One characteristic change is the shrinkage of the dendritic arbor, which, like the branches of a tree, make up most of a neuron's surface area. Indeed, loss of dendritic branches has been observed in people with AD as well as in AD mouse models (see Oct 2004 news story and Grutzendler et al., 2007). In addition to these morphological changes, neuronal activity changes as disease progresses, and some neurons become more excitable (see Sep 2008 news story and May 2012 news story). However, understanding the relationship between morphological deterioration and changes to neuronal activation patterns has been difficult.
To connect the dots, Remy, an electrophysiologist, invoked a basic law of physics. “If you reduce the surface area of the cell membrane, you need less current to charge the membrane and trigger the neuron to fire,” he said. Remy hypothesized that the loss of dendritic branches in neurodegenerative disease would therefore boost neuronal excitability.
To test this idea, first author Zuzana Šišková and colleagues turned to an APP/PS1 mouse model (see Willuweit et al., 2009). The researchers first measured the excitability of hippocampal neurons using an in vivo whole-cell patch-clamp technique: They inserted electrodes into the CA1 pyramidal layer of the hippocampus of deeply anesthetized mice and recorded neuronal firing patterns. They found that compared to neurons in wild-type or young APP/PS1 mice, neurons in plaque-ridden 10- to 14-month-old APP/PS1 mice more often fired spontaneously. They tended to go overboard, firing bursts of back-to-back action potentials rather than just one at a time. This hyperactivity increased whole network bursts, as detected by measuring local field potentials in the hippocampus.
The network bursts look like those that trigger epilepsy, commented Igor Timofeev of the University of Laval in Quebec. If confirmed, these findings would jibe with observations that some young AD patients have epileptic seizures, he wrote to Alzforum.
To more characterize more precisely what the neurons were doing, the researchers turned to hippocampal slice cultures. As with the in vivo recordings, neurons in slices from APP/PS1 mice fired more often than wild-type, and in bursts rather than single-action potentials. APP/PS1 neurons also required less current to trigger an action potential. To find out if this hyperexcitability correlated with changes in the neuron’s shape or size, the researchers injected biocytin—a dye that reveals dendritic branch structure—into the cells while recording. Fluorescence imaging showed that dendritic branches were 25 percent shorter and there were 22 percent fewer of them in old APP/PS1 mice than in wild-type or younger APP/PS1 mice without plaques. Overall, old APP/PS1 mice lost 20 percent of their dendritic surface area. Given that dendrites make up most of a CA1 pyramidal neuron’s surface area, these alterations had a big impact on the total size of the cells. When the researchers examined neurons with a high-resolution fluorescent-imaging technique called stimulated emission depletion (STED), they found fewer dendritic spines in APP/PS1 neurons (see Apr 2006 news story). The loss was most pronounced at the apical tuft dendritic branches, which are furthest from the soma. Any loss of spines, Remy said, decreases the total surface area of the neuron. The correlation between surface area lost and hyperactivity gained suggested to Remy that the two processes were intertwined in the context of amyloid plaque destruction.
To cinch the connection, Šišková and colleagues modeled the effects of dendrite loss with the computer program NEURON, which simulates electrical properties of cell networks. Each “neuron” in the model is made up of tens of thousands of tiny cylinders that can propagate electrical signals. Researchers can plug in parameters such as the size, number, and surface area of “dendritic branches” into the program, and simulate changes in the excitability of the “cell.” Based on the loss of dendritic structure seen in their morphological data, the researchers modeled 30 hypothetical neurons from APP/PS1 mice. They also created an in silico wild-type CA1 neuron for comparison. As with their data on real cells, the computational model predicted that the APP/PS1 neurons would fire more frequently, and would tend to fire spontaneously in bursts. When the researchers simulated the electrical stimulation, the model predicted that the APP/PS1 neurons would require less current to fire than the wild-type neuron.
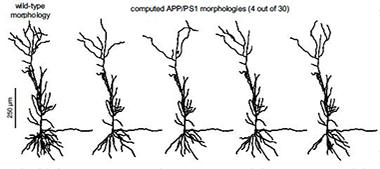
Mix and Match Branches.
A computer model generated hypothetical pyramidal neurons with either wild-type (left) or APP/PS1 (four on right) dendritic branch structures. The neurons on the right have smaller and fewer branches and thus different electrical properties. [Image courtesy of Zuzana Šišková et al., Neuron 2014.]
The researchers also simulated a more physiological scenario in which only some dendrites received signals that then propagated to the cell body, as opposed to delivering an overall current to the cell. Normally, neurons “hear” signals from their outermost branches less well than they do those from close to the soma because signals peter out as they travel, Remy said. When he simulated this type of distant stimulation, he found that excitatory post synaptic potentials (EPSPs) traveled more readily to the soma of the cells in APP/PS1 neurons.
The researchers next simulated the effect of rhythmic cumulative activation of 200 to 250 synapses at once, which resembles an input pattern CA1 neurons receive in vivo during behavior. This activation resulted in dramatic burst firing. “The neurons basically yell louder,” Remy said.
The modelling supported the idea that a simpler dendritic architecture sensitizes neurons to stimulation. Remy said more research is needed to determine whether dendritic degeneration and neuronal hyperexcitability are the cause or a consequence of network connectivity changes that occur in AD. Remy said one possible consequence of revved-up neuronal firing is the sapping of resources, adding that over time, this could tax neuronal circuits and exacerbate neurodegeneration.
Is the structure-function relationship specific to Aβ pathology, or a side effect of APP overexpression? Remy’s group did not use APP processing inhibitors to isolate the role of Aβ. However, he said, the problems occurred in mice after plaques appeared. Furthermore, the purpose of his study was not to point a finger at a specific species of Aβ or tau as the cause of neurodegeneration, but rather to uncover a fundamental relationship between degeneration and neuronal activity, he said. It is possible that such a relationship exists in other neurodegenerative diseases, even if they have different causes, Remy said. —Jessica Shugart
References
News Citations
- Caught in the Act—Amyloid Damages Neurons
- Hyperactive Neurons and Amyloid, Side by Side
- Soluble Aβ Takes Blame for Hyperactive Neurons in Mouse Brain
- Kiss and Tell—STED Microscopy Resolves Vesicle Recycling Question
Paper Citations
- Grutzendler J, Helmin K, Tsai J, Gan WB. Various dendritic abnormalities are associated with fibrillar amyloid deposits in Alzheimer's disease. Ann N Y Acad Sci. 2007 Feb;1097:30-9. PubMed.
- Willuweit A, Velden J, Godemann R, Manook A, Jetzek F, Tintrup H, Kauselmann G, Zevnik B, Henriksen G, Drzezga A, Pohlner J, Schoor M, Kemp JA, von der Kammer H. Early-onset and robust amyloid pathology in a new homozygous mouse model of Alzheimer's disease. PLoS One. 2009;4(11):e7931. PubMed.
Further Reading
Papers
- Spires TL, Hyman BT. Neuronal structure is altered by amyloid plaques. Rev Neurosci. 2004;15(4):267-78. PubMed.
Primary Papers
- Šišková Z, Justus D, Kaneko H, Friedrichs D, Henneberg N, Beutel T, Pitsch J, Schoch S, Becker A, von der Kammer H, Remy S. Dendritic structural degeneration is functionally linked to cellular hyperexcitability in a mouse model of Alzheimer's disease. Neuron. 2014 Dec 3;84(5):1023-33. Epub 2014 Nov 13 PubMed.
Annotate
To make an annotation you must Login or Register.
Comments
Universite Laval
The key finding in this paper is a reduction of the dendritic tree and some reduction of spines in about 1-year-old APP/PS1 mice. At least some studies suggest that this particular mouse is a good model of Alzheimer disease. Similar reduction in dendritic volume was found in patients with several neurodegenerative disorders, including Alzheimer disease.
The next finding of the paper is predictable from neuronal cable theory, i.e., that smaller cellular volume (or dendritic volume) equates to higher neuronal excitability. The authors provide references in the introduction on these studies. The established theoretical framework, however, needed experimental confirmation and the present study shows convincingly that indeed the intrinsic excitability of neurons is increased in hippocampal CA1 pyramidal neurons from APP/PS1 mice.
The next finding of the study, which was much less predicted from theory, is that anesthetized 1-year-old APP/PS1 mice display bursts of network activities. The nature of these bursts is unclear. There are at least two possibilities. One is that APP/PS1 mice have different sensitivity to anesthesia, and therefore these burst are appropriate to some anesthesia-related phenomena. The other possibility is more exciting. Are these bursts paroxysmal? Are they a signature of epileptic discharges? The shown spectrogram (Fig. 1 D) suggests that these bursts of network activity can be epileptiform.
Indeed, several studies demonstrate that “young” Alzheimer patients (50 years old) have a high likelihood to develop epilepsy. Because 1-year-old mice more or less correspond to 40- to 50-year-old humans, they may reveal comorbidities related to the Alzheimer condition, namely epilepsy. To show this convincingly, we need in vivo behavioral and electrophysiological experiments on non-anesthetized mice that are 1 year and older.
View all comments by Igor TimofeevUniversity College London
In this elegant study, Šišková et al. used a combination of approaches to examine the morphological and electrophysiological properties of pyramidal neurons in the CA1 region of the hippocampus in a transgenic mouse model of Alzheimer's disease amyloidosis. They provide a morphological explanation for abnormal hippocampal hyperexcitability in mice with high plaque load and memory impairment.
By using in vivo whole-cell recordings, the team demonstrated that in 10- to 14-month-old APP/PS1 mice, CA1 neurons showed massively elevated firing rates and a frequent occurrence of action potential bursts when compared with age-matched wild-type mice. Recordings in slices confirmed and extended these in vivo results. Next, morphological analyses using confocal and super-resolution STED microscopy revealed that in the transgenic mice the dendrites of CA1 neurons had a reduced length and surface area as well as a reduced spine density. Subsequent modeling led the authors to conclude that this dendritic degeneration was sufficient to explain the hyperexcitability of CA1 neurons.
The study nicely confirms previous work, which demonstrated Aβ related hyperactivity of neuronal circuits (see, e.g., Palop et al., 2007; Busche et al., 2008; Busche et al., 2012), but adds the important insight that such hyperexcitability—at least in an advanced disease stage—could be driven directly by neurodegeneration. I believe that the finding of this structure-function relation is very exciting and represents a major advance in our understanding of how Aβ pathology alters brain function at the level of individual neurons. This novel mechanism may also have broad implications for other diseases that are associated with neurodegeneration.
The study raises a number of basic and disease-related questions. For instance, I noticed that in wild-type animals the CA1 neuronal firing frequency of ~3 Hz was much higher than that in previous studies reporting firing rates below 1 Hz under similar conditions (see, e.g., Misuzeki and Buzsaki, 2013; Grienberger et al., 2014). In addition, in the wild-type mice the team detected a surprisingly low number of action potential bursts in vivo (~7 percent) and after current injection in slices (~4 percent), while previous papers had reported a higher occurrence of bursting neurons (see, e.g., Graves et al., 2012). I wonder whether these differences are due to the older age of animals used by Šišková et al., which would be very interesting per se. Burst firing is widely regarded as a key property of hippocampal neurons and may serve important physiological functions, e.g., for synaptic plasticity (see, e.g., Lisman, 1997; Xu et al., 2012). Therefore, the paper should prompt further studies to more specifically evaluate the properties of bursting neurons in aging and in the disease state.
It would be very interesting to see whether the observed effects are specific to Aβ-related structural deficits or can be found in other models of neurodegeneration as well. Furthermore, I wonder whether improving the morphological abnormalities—as has been reported by the use of locally applied antibodies against Aβ (see, e.g., Lombardo et al., 2003; Brendza et al., 2005)—can rescue the functional deficits.
References:
Palop JJ, Chin J, Roberson ED, Wang J, Thwin MT, Bien-Ly N, Yoo J, Ho KO, Yu GQ, Kreitzer A, Finkbeiner S, Noebels JL, Mucke L. Aberrant excitatory neuronal activity and compensatory remodeling of inhibitory hippocampal circuits in mouse models of Alzheimer's disease. Neuron. 2007 Sep 6;55(5):697-711. PubMed.
Busche MA, Eichhoff G, Adelsberger H, Abramowski D, Wiederhold KH, Haass C, Staufenbiel M, Konnerth A, Garaschuk O. Clusters of hyperactive neurons near amyloid plaques in a mouse model of Alzheimer's disease. Science. 2008 Sep 19;321(5896):1686-9. PubMed.
Busche MA, Chen X, Henning HA, Reichwald J, Staufenbiel M, Sakmann B, Konnerth A. Critical role of soluble amyloid-β for early hippocampal hyperactivity in a mouse model of Alzheimer's disease. Proc Natl Acad Sci U S A. 2012 May 29;109(22):8740-5. Epub 2012 May 16 PubMed.
Mizuseki K, Buzsáki G. Preconfigured, skewed distribution of firing rates in the hippocampus and entorhinal cortex. Cell Rep. 2013 Sep 12;4(5):1010-21. Epub 2013 Aug 29 PubMed.
Grienberger C, Chen X, Konnerth A. NMDA receptor-dependent multidendrite Ca(2+) spikes required for hippocampal burst firing in vivo. Neuron. 2014 Mar 19;81(6):1274-81. Epub 2014 Feb 20 PubMed.
Graves AR, Moore SJ, Bloss EB, Mensh BD, Kath WL, Spruston N. Hippocampal pyramidal neurons comprise two distinct cell types that are countermodulated by metabotropic receptors. Neuron. 2012 Nov 21;76(4):776-89. PubMed.
Lisman JE. Bursts as a unit of neural information: making unreliable synapses reliable. Trends Neurosci. 1997 Jan;20(1):38-43. PubMed.
Xu W, Morishita W, Buckmaster PS, Pang ZP, Malenka RC, Südhof TC. Distinct neuronal coding schemes in memory revealed by selective erasure of fast synchronous synaptic transmission. Neuron. 2012 Mar 8;73(5):990-1001. PubMed.
Lombardo JA, Stern EA, McLellan ME, Kajdasz ST, Hickey GA, Bacskai BJ, Hyman BT. Amyloid-beta antibody treatment leads to rapid normalization of plaque-induced neuritic alterations. J Neurosci. 2003 Nov 26;23(34):10879-83. PubMed.
Brendza RP, Bacskai BJ, Cirrito JR, Simmons KA, Skoch JM, Klunk WE, Mathis CA, Bales KR, Paul SM, Hyman BT, Holtzman DM. Anti-Abeta antibody treatment promotes the rapid recovery of amyloid-associated neuritic dystrophy in PDAPP transgenic mice. J Clin Invest. 2005 Feb;115(2):428-33. PubMed.
View all comments by Marc Aurel BuscheMake a Comment
To make a comment you must login or register.