Do Platelets Provide a Platform for Aβ Aggregation?
Quick Links
Platelets are expert at patching up leaks in blood vessels, but could they go overboard and seed amyloid-β plaques as well? As reported in Science Signaling on May 24, Aβ monomers bind integrin receptors on human platelets in vitro, triggering the release of the chaperone clusterin, which then facilitates Aβ aggregation. Blocking platelet activation reduced cerebral amyloid angiopathy (CAA) in a mouse model of Alzheimer’s disease. Led by Margitta Elvers at Heinrich Heine University in Düsseldorf, Germany, the researchers proposed that anti-platelet therapy could one day do the same in people.
The findings add mechanistic oomph to previous reports implicating platelets in AD, commented Costantino Iadecola of Weill Cornell Medical College in New York. However, any future attempts to treat AD patients with anti-platelet therapy will be tempered by the potential for bleeding risks, Iadecola and other researchers warned.
Platelets use their anucleated bodies to patch blood vessel leaks—a line of defense that triggers the formation of blood clots. Multiple studies hint that platelets also play a role in AD. In dementia patients platelets seem to be more activated. People with brain emboli—teeny bits of clotting material floating in the brain vessels, a characteristic of overactive platelets—decline faster on cognitive tests than people without them (see Ciabattoni et al., 2007, and Feb 2012 news). Moreover, platelets are a major source of Aβ in the blood, where the peptide can reportedly activate them, triggering their adhesion to each other (see Roher et al., 2009; Bush et al., 1990; and Shen et al., 2008). Interestingly, platelets also affect Aβ; work from Elvers’ lab revealed that Aβ fibrillizes when added to platelets in vitro, and the researchers spotted platelets comingling with vascular amyloid plaques in APP23 mice (see Gowert et al., 2014; and Jarre et al., 2014).
For this study, first author Lili Donner and colleagues wanted to chip away at the mechanisms behind the two-way interactions between Aβ and platelets. As measured by immunofluorescence, Congo red staining, and electron microscopy, platelets incubated with monomeric Aβ40 for three days stimulated formation of Aβ fibrils. This fibrillization depended upon the presence of the platelets themselves, as simply mixing platelet supernatant with Aβ promoted no fibrils. Aβ also stimulated signaling within the platelets, as evidenced by elevated platelet aggregation and the activation of various signaling molecules as well as secretion of the chaperone clusterin (aka Apolipoprotein J). Genetic variants in this gene associate with AD (see Alzgene). Interestingly, platelets from clusterin knockout mice failed to promote Aβ aggregation, hinting that Aβ may stimulate the release of the very factor—clusterin—that triggers its aggregation on platelets. In keeping with this idea, adding clusterin protein to these cultures promoted Aβ aggregation.
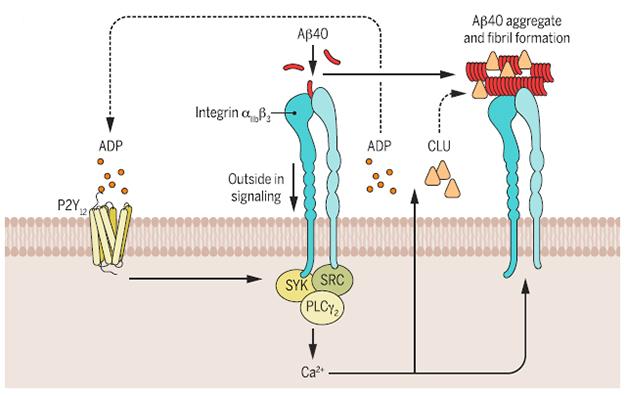
Fibrillization Platform. On platelets, Aβ monomers trigger the integrin receptor, which boosts release of ADP and clusterin, accelerating Aβ fibrillization. [Image courtesy of McFayden and Peter, Focus Sci Signaling 2016.]
How might Aβ trigger clusterin release? The researchers found that the peptide bound to the fibrinogen receptor integrin αIIbβ3, tripping off a signaling cascade resulting in the release of the apolipoprotein. Fibrinogen facilitates the adhesion of platelets to each other, and is also the precursor to fibrin, a major component of blood clots. Aβ also triggered the release of adenosine diphosphate from the platelets, which enhanced clusterin release through binding to the P2Y12 receptor on the platelet surface (see image above). The researchers found that blocking this purinergic receptor with the blood-thinning drug clopidogrel severely dampened both clusterin release from platelets and their penchant for aggregating Aβ.
The researchers next sought to strengthen the proposed connection between Aβ and integrin by measuring Aβ aggregation in the presence of platelets from people who have Glanzmann’s thrombasthenia, a rare blood-clotting disorder caused by poor expression of the αIIbβ3 integrin on platelets. Platelets from patients with the most severe form of GT failed to promote Aβ fibrillization; however, fibrils did form when platelets from GT patients expressed a moderate level of this particular integrin.
To test if platelets contribute to CAA in vivo, the researchers treated 13-month-old APP23 mice for three months with clopidogrel to reduce platelet activation and prevent clusterin release and Aβ aggregation. While the treated and untreated mice had similar burdens of Aβ plaques in the brain parenchyma, the burden of vascular plaque was only half in the treated mice. This suggested that blocking platelet function slowed vascular amyloid deposition.
These results seemingly conflict with those from John Fryer’s lab at the Mayo Clinic in Jacksonville, Florida. His group recently reported that compared to APP/PS1 mice, those lacking clusterin had more vascular amyloid, suggesting that clusterin prevented, rather than promoted, vascular amyloid deposition (see Nov 2015 conference news).
Elvers proposes anti-platelet therapy as a potential treatment strategy for CAA. The vascular amyloid-specific action of clopidogrel provides support for this idea, commented Ilaria Canobbio of the University of Pavia in Italy, whose previous work implicated Aβ in platelet activation (see Canobbio et al., 2014). However, such drugs are only used under acute conditions such as myocardial infarction to prevent clotting, and pose substantial bleeding risks, Canobbio added. Clopidogrel’s action is also short-lived, as the active metabolite of the compound has a half-life of less than one hour.
“Clopidogrel is typically not considered for CAA or AD. Thus, from a therapeutic standpoint, it could prove important to prevent amyloid deposition in blood vessel walls, which could in turn reduce the complications of the angiopathy,” commented Marwan Sabbagh of Barrow Neurological Institute in Phoenix. “Ironically, this is counterintuitive because we typically avoid anti-platelet treatments in hemorrhagic events such as CAA.”
Newer therapies, such as antibodies that block the integrin receptor without activating it, could potentially prevent Aβ aggregation with a longer half-life, commented James McFadyen and Karlheinz Peter of Monash University in Melbourne, Australia, in an editorial (see Schwarz et al., 2006; McFadyen and Peter, 2016). The pharmacokinetic profiles of these therapies would provide some wiggle room in the extent to which they block platelet activation, thus potentially reducing the bleeding risk, they pointed out.
McFadyen and Peter wondered whether platelets could exacerbate other diseases, including diabetes and atherosclerosis, which are characterized by deposition of different types of protein along blood vessel walls. “In light of the data presented here, it is tempting to speculate that platelet activation in this context may also establish a self-perpetuating cycle that precipitates the formation of further aggregates of misfolded proteins, thereby directly promoting the progression of disease,” they wrote.
Iadecola thought Elvers’ results dovetailed nicely with previous data from his lab, which revealed that CD36, a scavenger receptor on platelets, promoted vascular amyloid deposition in mice (see Park et al., 2013). He noted that the Aβ aggregation via platelets could at least partly explain why people with AD have elevated levels of atherosclerosis in brain blood vessels, since Aβ fibrils may contribute to sticky platelet clots that serve as a scaffold for atherosclerotic plaques.
The most intriguing unanswered question, Iadecola said, is how and where platelets and Aβ meet up and promote CAA. “What doesn’t make sense is how platelets decorated with Aβ cross the endothelium and get into the smooth muscle cells where CAA accumulates,” he said. William Van Nostrand of Stony Brook University in New York stressed the same quandary. “Perhaps platelets play a role later in the disease when there is compromise of vascular integrity and exposure to platelet components,” he suggested (see full comment below).
Furthermore, whether the source of Aβ for the platelet fibrils comes from the brain or from the platelets themselves is up for debate. Van Nostrand pointed out that while strong evidence exists for the role of neuronal Aβ in vascular amyloid deposition, platelets process APP primarily via the non-amyloidogenic α-secretase pathway, producing only small amounts of Aβ40. Elvers and colleagues had to use concentrations of Aβ40 far exceeding that found in the blood, he added. Others also noted this caveat, and lamented the small number of animals used in some experiments.—Jessica Shugart
References
News Citations
- Silent Vascular Disease May Hasten Dementia Progression
- Alzheimer’s Risk Genes Give Up Some Secrets at SfN
Research Models Citations
Alzpedia Citations
Paper Citations
- Ciabattoni G, Porreca E, Di Febbo C, Di Iorio A, Paganelli R, Bucciarelli T, Pescara L, Del Re L, Giusti C, Falco A, Sau A, Patrono C, Davì G. Determinants of platelet activation in Alzheimer's disease. Neurobiol Aging. 2007 Mar;28(3):336-42. PubMed.
- Roher AE, Esh CL, Kokjohn TA, Castaño EM, Van Vickle GD, Kalback WM, Patton RL, Luehrs DC, Daugs ID, Kuo YM, Emmerling MR, Soares H, Quinn JF, Kaye J, Connor DJ, Silverberg NB, Adler CH, Seward JD, Beach TG, Sabbagh MN. Amyloid beta peptides in human plasma and tissues and their significance for Alzheimer's disease. Alzheimers Dement. 2009 Jan;5(1):18-29. PubMed.
- Bush AI, Martins RN, Rumble B, Moir R, Fuller S, Milward E, Currie J, Ames D, Weidemann A, Fischer P. The amyloid precursor protein of Alzheimer's disease is released by human platelets. J Biol Chem. 1990 Sep 15;265(26):15977-83. PubMed.
- Shen MY, Hsiao G, Fong TH, Chen HM, Chou DS, Lin CH, Sheu JR, Hsu CY. Amyloid beta peptide-activated signal pathways in human platelets. Eur J Pharmacol. 2008 Jul 7;588(2-3):259-66. PubMed.
- Gowert NS, Donner L, Chatterjee M, Eisele YS, Towhid ST, Münzer P, Walker B, Ogorek I, Borst O, Grandoch M, Schaller M, Fischer JW, Gawaz M, Weggen S, Lang F, Jucker M, Elvers M. Blood platelets in the progression of Alzheimer's disease. PLoS One. 2014;9(2):e90523. Epub 2014 Feb 28 PubMed.
- Jarre A, Gowert NS, Donner L, Münzer P, Klier M, Borst O, Schaller M, Lang F, Korth C, Elvers M. Pre-activated blood platelets and a pro-thrombotic phenotype in APP23 mice modeling Alzheimer's disease. Cell Signal. 2014 Sep;26(9):2040-50. Epub 2014 Jun 11 PubMed.
- Canobbio I, Guidetti GF, Oliviero B, Manganaro D, Vara D, Torti M, Pula G. Amyloid β-peptide-dependent activation of human platelets: essential role for Ca2+ and ADP in aggregation and thrombus formation. Biochem J. 2014 Sep 15;462(3):513-23. PubMed.
- Schwarz M, Meade G, Stoll P, Ylanne J, Bassler N, Chen YC, Hagemeyer CE, Ahrens I, Moran N, Kenny D, Fitzgerald D, Bode C, Peter K. Conformation-specific blockade of the integrin GPIIb/IIIa: a novel antiplatelet strategy that selectively targets activated platelets. Circ Res. 2006 Jul 7;99(1):25-33. Epub 2006 Jun 15 PubMed.
- McFadyen J, Peter K. Forget about thrombosis: Platelets and Alzheimer's disease, yet another sticky situation. Sci Signal. 2016 May 24;9(429):fs9. PubMed.
- Park L, Zhou J, Zhou P, Pistick R, El Jamal S, Younkin L, Pierce J, Arreguin A, Anrather J, Younkin SG, Carlson GA, McEwen BS, Iadecola C. Innate immunity receptor CD36 promotes cerebral amyloid angiopathy. Proc Natl Acad Sci U S A. 2013 Feb 19;110(8):3089-94. PubMed.
External Citations
Further Reading
Papers
- Hohmann JD, Peter K. Activated-platelet targeting of CD39 as a potential way forward. The quest for efficient antithrombotic therapy without associated bleeding complications. Hamostaseologie. 2016 Feb 10;36(1):17-25. Epub 2015 Sep 2 PubMed.
- Canobbio I, Abubaker AA, Visconte C, Torti M, Pula G. Role of amyloid peptides in vascular dysfunction and platelet dysregulation in Alzheimer's disease. Front Cell Neurosci. 2015;9:65. Epub 2015 Mar 3 PubMed.
Primary Papers
- Donner L, Fälker K, Gremer L, Klinker S, Pagani G, Ljungberg LU, Lothmann K, Rizzi F, Schaller M, Gohlke H, Willbold D, Grenegard M, Elvers M. Platelets contribute to amyloid-β aggregation in cerebral vessels through integrin αIIbβ3-induced outside-in signaling and clusterin release. Sci Signal. 2016 May 24;9(429):ra52. PubMed.
Annotate
To make an annotation you must Login or Register.
Comments
Barrow Neurological Institute
This research by Donner and colleagues makes important contributions in understanding mechanistically how amyloid is deposited in cerebral amyloid angiopathy. It appears that platelets play an important role in Aβ fibrillization. Specifically, amyloid appears to bind to integrin, which stimulates ADP and clusterin secretion from platelets. The clusterin then promotes amyloid aggregation, which leads to accumulation. This explains, in essence, how amyloid is bound to vessel walls.
Also important to this observation is the evidence that clopidogrel could reduce this process by inhibiting clusterin release. Clopidogrel is typically not considered for CAA or AD. Thus, from a therapeutic standpoint, it could prove important to prevent amyloid deposition in blood vessel walls, which could in turn reduce the complications of the angiopathy. Ironically, this is counterintuitive because we typically avoid anti-platelet treatments in hemorrhagic events such as CAA.…More
University of Rhode Island
This recent article of Donner et al. provides an elegant series of largely ex vivo experiments to demonstrate how soluble Aβ peptide can activate cultured platelets, cause release of the Aβ chaperone clusterin, and promote Aβ aggregation. These processes were shown to be mediated by the RHDS motif of Aβ interacting with the platelet surface integrin α11bβ3, stimulating an outside-in signaling cascade that facilitates clusterin release and Aβ aggregation. Interestingly, platelets from patients with Glanzman thrombasthenia (GT), which have markedly reduced levels of dysfunctional α11bβ3, were deficient in clusterin release and in promotion of Aβ aggregation, further connecting these two events in these experiments. The most intriguing aspect of the work involved studies with APP23 transgenic mice that overexpress human AβPP and develop both brain parenchymal and vascular amyloid deposition. The authors show that treatment of APP23 mice with the anti-platelet drug clopidogrel appears to reduce the number of vessels with amyloid deposits and the adherence of platelets to these vessels. These findings provide evidence that platelets may contribute to the development of cerebral vascular amyloid formation and could provide a target to treat the condition of cerebral amyloid angiopathy.…More
The notion that platelets could play a role in Alzheimer’s disease pathology has been considered for more than 25 years now, following the initial discovery that platelets contain very high levels of AβPP (Van Nostrand et al., 1990; Bush et al., 1990) and can release primarily Aβ40 peptide upon activation, although these levels are quite low at the pg/ml level (Li et al., 1998; Casoli et al., 2007). Clusterin has been recognized as an important chaperone for Aβ that can promote its fibrillar assembly, and clusterin absence dramatically reduces amyloid plaque formation in human AβPP transgenic mice (Demattos et al., 2002). The present study ties these previous findings together, providing a potential mechanism as to how platelets can facilitate cerebral vascular amyloid formation.
As intriguing as these findings are, they raise a number of key follow-up questions to ascertain the importance of this mechanism to cerebral vascular amyloid formation. First, what is the source of Aβ that can promote such interactions with platelets and lead to vascular amyloid formation? As mentioned above, although platelets contain very high levels of AβPP, the vast majority of this appears to be processed by the non-amyloidogenic α-secretase pathway. Therefore, platelets seem to contain very low (pg) amounts of Aβ. The studies presented in this article required micromolar concentrations of Aβ40 to promote stimulation of platelets and aggregation of Aβ. On the other hand, there is strong evidence, particularly with transgenic mouse models, that in the brain neuronal production of Aβ causes cerebral vascular amyloid formation (Calhoun et al., 1998; Davis et al., 2004). Further, vascular amyloid formation occurs on the abluminal side of cerebral capillaries and in the tunica media of small arterioles. Thus, the question as to how the initial seeding and expansion of abluminal vascular amyloid could be initiated by circulating platelets needs to be addressed. Perhaps platelets play a role later in the disease when there is compromise of vascular integrity and exposure to platelet components.
Second, although the most common form of cerebral amyloid angiopathy (CAA) involves Aβ, numerous other amyloidogenic proteins can cause this condition, including prion proteins, cystation C, amyloid British protein, amyloid Danish protein, gelsolin, and others. Since these amyloid proteins do not harbor the RHDS motif of Aβ, which appears to be important for promoting the platelet responses, are markedly different mechanisms involved in these CAA disorders? Also, secreted sAβPPα, which harbors the “RHDS” motif at its C-terminal end, is released at very high levels from activated platelets. Can this species of sAβPPα also interact with the platelet surface integrin α11bβ3 to promote platelet outside-in signaling and clusterin release? Can it block Aβ interactions with platelets and Aβ aggregation?
Finally, the authors suggest that anti-platelet therapy may be useful for treating CAA. As in most cases, this type of approach is a double-edged sword. Indeed, CAA can promote small vessel occlusions, which clog up with platelets as the authors previously showed. In this case, preventing the adherence and clumping of platelets could be helpful. However, CAA can also cause loss of vessel integrity and hemorrhage. In this case, preventing platelets from serving their normal function in forming a thrombotic plug would be counterproductive and prolong detrimental bleeding into the brain. Although these are significant questions that need to be addressed in future studies, the present article continues to support the important concept of vascular contributions to Alzheimer’s disease.
References:
Bush AI, Martins RN, Rumble B, Moir R, Fuller S, Milward E, Currie J, Ames D, Weidemann A, Fischer P. The amyloid precursor protein of Alzheimer's disease is released by human platelets. J Biol Chem. 1990 Sep 15;265(26):15977-83. PubMed.
Calhoun ME, Burgermeister P, Phinney AL, Stalder M, Tolnay M, Wiederhold KH, Abramowski D, Sturchler-Pierrat C, Sommer B, Staufenbiel M, Jucker M. Neuronal overexpression of mutant amyloid precursor protein results in prominent deposition of cerebrovascular amyloid. Proc Natl Acad Sci U S A. 1999 Nov 23;96(24):14088-93. PubMed.
Casoli T, Di Stefano G, Giorgetti B, Grossi Y, Balietti M, Fattoretti P, Bertoni-Freddari C. Release of beta-amyloid from high-density platelets: implications for Alzheimer's disease pathology. Ann N Y Acad Sci. 2007 Jan;1096:170-8. PubMed.
Davis J, Xu F, Deane R, Romanov G, Previti ML, Zeigler K, Zlokovic BV, Van Nostrand WE. Early-onset and robust cerebral microvascular accumulation of amyloid beta-protein in transgenic mice expressing low levels of a vasculotropic Dutch/Iowa mutant form of amyloid beta-protein precursor. J Biol Chem. 2004 May 7;279(19):20296-306. Epub 2004 Feb 25 PubMed.
Demattos RB, O'Dell MA, Parsadanian M, Taylor JW, Harmony JA, Bales KR, Paul SM, Aronow BJ, Holtzman DM. Clusterin promotes amyloid plaque formation and is critical for neuritic toxicity in a mouse model of Alzheimer's disease. Proc Natl Acad Sci U S A. 2002 Aug 6;99(16):10843-8. PubMed.
Li QX, Whyte S, Tanner JE, Evin G, Beyreuther K, Masters CL. Secretion of Alzheimer's disease Abeta amyloid peptide by activated human platelets. Lab Invest. 1998 Apr;78(4):461-9. PubMed.
Van Nostrand WE, Schmaier AH, Farrow JS, Cunningham DD. Protease nexin-II (amyloid beta-protein precursor): a platelet alpha-granule protein. Science. 1990 May 11;248(4956):745-8. PubMed.
University of Pavia
Aβ peptides accumulate in cerebral vessels, causing cerebral amyloid angiopathy (CAA), and in brain parenchyma correlating with Alzheimer’s disease (AD). It is known that besides neurons, circulating platelets contain high amount of Aβ peptides. Upon activation, platelets release Aβ in the circulation, which in turn reinforce platelet adhesion, activation, and aggregation. Platelets are also able to mobilize soluble Aβ peptides into fibrillary Aβ and induce fibrillar Aβ aggregate formation in culture.
In this recent paper in Science Signaling, the group of Margitta Elvers demonstrates that soluble Aβ40, via its RHDS sequence, is able to bind integrin αIIbβ3. This binding induces platelet adhesion and integrin αIIbβ3 outside-in activation and promotes release of clusterin and ADP. Clusterin is a chaperone that is associated with the severity of AD and influences the structure and toxicity of Aβ peptides, contributing to the formation Aβ fibrils. ADP is a platelet agonist. We have previously demonstrated that it plays a crucial role in Aβ peptide-induced platelet activation (Canobbio et al., 2014). Here the authors confirm our observation and demonstrated that ADP is also important in platelet-mediated fibril formation. All these data point to a pivotal role of platelets and ADP in Aβ deposition and oligomerization in cerebral vessels. More interestingly, the authors demonstrate that treating AD mice (APP23) with the ADP receptor P2Y12 antagonist clopidogrel (commonly used in anti-platelet therapies), inhibits platelet activation and clusterin release and decreases CAA. These results open new perspectives on novel therapeutic approaches to prevent Aβ fibril formation and CAA development. …More
References:
Canobbio I, Guidetti GF, Oliviero B, Manganaro D, Vara D, Torti M, Pula G. Amyloid β-peptide-dependent activation of human platelets: essential role for Ca2+ and ADP in aggregation and thrombus formation. Biochem J. 2014 Sep 15;462(3):513-23. PubMed.
Make a Comment
To make a comment you must login or register.