Design From Disorder: New Test Reveals Different Strains of SOD1
Quick Links
When individual proteins form aggregates, they shed the shapes they once took as lone monomers for others that fit in with the collective. A new technique capitalizes on this often-disorderly transition to detect different strains of aggregated SOD1, a protein that forms inclusions associated with amyotrophic lateral sclerosis (ALS). Described March 23 in Proceedings of the National Academy of Sciences, the method employs an arsenal of antibodies to detect regions of the protein only exposed when it assumes non-native structures. Researchers led by Stefan Marklund at Umea University in Sweden correlated one strain of SOD1 with the rate of disease progression, and plan to use the technique to identify SOD1 strains in humans next.
“This is a well-executed study, and it offers a reasonable approach to detecting different strains of aggregated proteins and studying their relationship with disease,” commented David Teplow of the University of California, Los Angeles, who was not involved in the work. “This technique could certainly be applied much more broadly than SOD1, to any of the amyloidogenic proteins,” he added.
Neil Cashman of the University of British Columbia in Vancouver commented that the paper adds SOD1 to the ranks of neurodegenerative proteins that exhibit strain behavior.
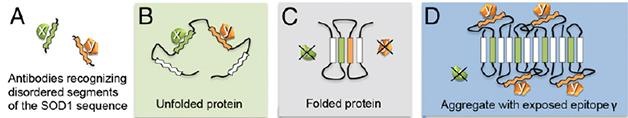
Epitope Mapping. Some antibodies raised against isolated fragments of SOD1 don’t see their targets when the protein is folded normally. However, some of those segments are exposed when the protein forms an aggregate. [Image courtesy of Bergh et al., PNAS 2015.]
The formation of toxic aggregates and smaller oligomers is a common feature of many neurodegenerative disorders, including ALS, Alzheimer's, and Parkinson's. Overwhelming evidence now suggests that there are different “strains” of Aβ, tau, α-synuclein, and even SOD1, and that these strains may spread from one cell to another (see Sep 2013 news; Clavaguera et al., 2013; Aug 2013 conference news; Bousset et al., 2013; Ayers et al., 2014). However, the detection and characterization of strains remain major challenges for the field.
First author Johan Bergh and colleagues went on a hunt for SOD1 aggregates using a novel approach they called binary epitope mapping. They developed a panel of antibodies that would only detect epitopes that are normally buried in the native protein, but that are exposed when it unravels. They theorized that some of those epitopes might also be exposed when the protein misfolds, as it does when forming aggregates. The researchers raised polyclonal antibodies against short peptide segments that spanned 90 percent of the SOD1 protein sequence. They found eight antibodies that detected full length, denatured SOD1, but not the natively folded form, indicating that the epitopes recognized were obscured within the protein’s folds (see image above).
The researchers tested the antibodies on spinal cord extracts from lines of mice overexpressing normal or mutant human SOD1. Four of the eight antibodies reacted to spinal cord extracts from G93A, G85R, and wild-type SOD1-expressing mice. The researchers dubbed these SOD1 aggregates “strain A.” In addition to strain A aggregates, D90A SOD1 mice contained others, designated “strain B," that lit up a different subset of the antibodies. The brain and spinal cords of each mouse contained the same strain, although there was much less in the brain. A third distinctive pattern emerged when the researchers generated SOD1 fibrils in vitro. Together, these findings suggested that something about the in vivo environment, and/or the nature of the mutation, dictated which strains of aggregates would form. They also questioned the reliance on in vitro aggregates as a model of those occurring during disease.

Signature of a Strain.
SOD aggregates in mouse models of ALS (top two panels), and one formed in vitro (bottom panel) bind antibodies that recognize different sequences (abscissa) of the protein, indicating their secondary structures expose unique sets of epitopes. [Image courtesy of Bergh et al., PNAS 2015.]
Teplow was struck by the clear difference in SOD1 strains that formed in the mice versus in vitro. Researchers struggle with such heterogeneity when they study Aβ as well, he said. Scientists often use Aβ oligomers and aggregates formed in test tubes without knowing how relevant they are to physiology (see Alzforum Webinar and Nature editorial). An assay like this could help determine if differences exist between aggregates whipped up on the bench versus those found in the brain, said Teplow.
The researchers next compared the stability of the aggregate strains. They exposed spinal cord extracts from symptomatic D90A mice to increasing concentrations of the powerful denaturant guanidine chloride, blasted them with sonication, and then captured any surviving SOD1 aggregates with a filter. This predominantly captured strain A, suggesting that strain B aggregates more readily fell apart. This finding hinted to the researchers that strain B could be more pathogenic, as previous studies had reported that fragile strains of Aβ or prion proteins more readily seeded the growth of new aggregates than stable strains did (see Bett et al., 2012; Xue et al., 2010; and Colby and Prusiner, 2011).
How did these different strains alter the course of disease? The researchers found that D90A mice that died earliest harbored stain B aggregates in their spinal cords, whereas those that lived longest had only strain A. Mice with lifespans somewhere in the middle died with a mixture of the two strains. Mice with strain B also had an earlier disease onset and shorter disease duration, implicating the fragile strain of SOD1 aggregates in promoting a rapid demise. These results suggested that, much as different tau strains brew their own pathological stew, different SOD1 strains dictate the course of disease (see May 2014 news).
“One of the most interesting hypotheses put forward in this study is that the CNS modulates the development of predominant aggregate strains,” noted Claudio Gomes of the faculty of sciences at the University of Lisbon. He said that the epitope-binding assay could be useful in studying how different factors affect SOD1 aggregate structures.
Bergh plans to expand the collection of antibodies in the hope of detecting other strains of SOD1. They will also test the antibodies on postmortem samples from human ALS patients. He hopes the assay will be sensitive enough to detect aggregates in patient cerebrospinal fluid, which would allow possible correlations with prognosis and disease progression. Teplow agreed that this would be useful, and commented that the assay has yet to be optimized to its maximum potential, for example by amplifying antibody signals. “The assay is really good, but could be made even better,” he said.
The assay may stand a good chance at detecting low levels of aggregates. In ALS mice, the SOD1 aggregates exist at 100μg/g wet weight, and the researchers reported that their binary epitope assay detected 1,000 times less. The heightened sensitivity of the assay is due to its combinatorial nature, as it detects a series of linear epitopes that define each strain rather than only a single three-dimensional structure, said Bergh.
Bergh said the ultimate goal is to use aggregate-specific antibodies as immunotherapy in ALS patients. Gomes agreed that the approach was a viable one. “Even if we don’t succeed in completely abolishing the formation of protein deposits, targeting and removal from the CNS of even a relatively small fraction of the most deleterious strains may result in an important delay in the emergence of severe disease,” he said.—Jessica Shugart
References
News Citations
- Does Aβ Come In Strains? Glimpse Into Human Brain Suggests Yes
- Are Protein Strains The Cause of Different Tauopathies?
- Like Prions, Tau Strains Are True to Form
Webinar Citations
Paper Citations
- Clavaguera F, Akatsu H, Fraser G, Crowther RA, Frank S, Hench J, Probst A, Winkler DT, Reichwald J, Staufenbiel M, Ghetti B, Goedert M, Tolnay M. Brain homogenates from human tauopathies induce tau inclusions in mouse brain. Proc Natl Acad Sci U S A. 2013 Jun 4;110(23):9535-40. PubMed.
- Bousset L, Pieri L, Ruiz-Arlandis G, Gath J, Jensen PH, Habenstein B, Madiona K, Olieric V, Böckmann A, Meier BH, Melki R. Structural and functional characterization of two alpha-synuclein strains. Nat Commun. 2013;4:2575. PubMed.
- Ayers JI, Fromholt S, Koch M, DeBosier A, McMahon B, Xu G, Borchelt DR. Experimental transmissibility of mutant SOD1 motor neuron disease. Acta Neuropathol. 2014 Dec;128(6):791-803. Epub 2014 Sep 28 PubMed.
- Bett C, Joshi-Barr S, Lucero M, Trejo M, Liberski P, Kelly JW, Masliah E, Sigurdson CJ. Biochemical properties of highly neuroinvasive prion strains. PLoS Pathog. 2012 Feb;8(2):e1002522. PubMed.
- Xue WF, Hellewell AL, Gosal WS, Homans SW, Hewitt EW, Radford SE. Fibril fragmentation enhances amyloid cytotoxicity. J Biol Chem. 2009 Dec 4;284(49):34272-82. PubMed.
- Colby DW, Prusiner SB. Prions. Cold Spring Harb Perspect Biol. 2011 Jan 1;3(1):a006833. PubMed.
External Citations
Further Reading
No Available Further Reading
Primary Papers
- Bergh J, Zetterström P, Andersen PM, Brännström T, Graffmo KS, Jonsson PA, Lang L, Danielsson J, Oliveberg M, Marklund SL. Structural and kinetic analysis of protein-aggregate strains in vivo using binary epitope mapping. Proc Natl Acad Sci U S A. 2015 Apr 7;112(14):4489-94. Epub 2015 Mar 23 PubMed.
Annotate
To make an annotation you must Login or Register.
Comments
UBC
This is a very interesting paper, which furthers the concept of "SOD1 strains" in ALS and mouse models of the disease. Infectious prions (CJD in humans, scrapie in sheep, BSE in cattle, and CWD in deer and elk) do not exist in one monolithic form, but rather can exhibit “strain behavior” in a particular species. In sheep scrapie, the first infectious prion passaged to mouse models, approximately 20 strains were cloned and defined on the basis of incubation time, brain regions predominantly affected, and biochemical features such as glycosylation preferences. It turns out that non-infectious prion-like agents causing common neurodegenerative diseases also exhibit strain behavior. For Alzheimer’s disease, strain properties of aggregated Aβ in human brains have been found to correlate with rate of progression of disease (Cohen et al., 2015). In Parkinson’s disease and in multiple systems atrophy, different strains of aggregated α-synuclein have been identified with different pathological activity on the aggregation of tau protein in cell lines in vitro (Guo et al., 2013) and on the vulnerable neuronal pool in human brain (Watts et al., 2013). Tauopathy has been found to propagate in at least two different strains (Ayers et al., 2014) that correspond to the clinical features and rate of progression in humans. And finally, at the end of last year, ALS SOD1 propagation joined the "strain concept" with the first transmissions of disease to a mouse model (Sanders et al., 2014)—G93A "seeds" transmitted differently than human in wild-type transgenic mice. Now the current article shows with great rigor that the propagating misfolding of D90A SOD1 in transgenic mice differs with regard to epitope exposure to G93A, G85R, and human wild-type SOD1.
References:
Cohen ML, Kim C, Haldiman T, ElHag M, Mehndiratta P, Pichet T, Lissemore F, Shea M, Cohen Y, Chen W, Blevins J, Appleby BS, Surewicz K, Surewicz WK, Sajatovic M, Tatsuoka C, Zhang S, Mayo P, Butkiewicz M, Haines JL, Lerner AJ, Safar JG. Rapidly progressive Alzheimer's disease features distinct structures of amyloid-β. Brain. 2015 Apr;138(Pt 4):1009-22. Epub 2015 Feb 15 PubMed.
Guo JL, Covell DJ, Daniels JP, Iba M, Stieber A, Zhang B, Riddle DM, Kwong LK, Xu Y, Trojanowski JQ, Lee VM. Distinct α-synuclein strains differentially promote tau inclusions in neurons. Cell. 2013 Jul 3;154(1):103-17. PubMed.
Watts JC, Giles K, Oehler A, Middleton L, Dexter DT, Gentleman SM, DeArmond SJ, Prusiner SB. Transmission of multiple system atrophy prions to transgenic mice. Proc Natl Acad Sci U S A. 2013 Nov 26;110(48):19555-60. Epub 2013 Nov 11 PubMed.
Sanders DW, Kaufman SK, DeVos SL, Sharma AM, Mirbaha H, Li A, Barker SJ, Foley AC, Thorpe JR, Serpell LC, Miller TM, Grinberg LT, Seeley WW, Diamond MI. Distinct tau prion strains propagate in cells and mice and define different tauopathies. Neuron. 2014 Jun 18;82(6):1271-88. Epub 2014 May 22 PubMed.
Ayers JI, Fromholt S, Koch M, DeBosier A, McMahon B, Xu G, Borchelt DR. Experimental transmissibility of mutant SOD1 motor neuron disease. Acta Neuropathol. 2014 Dec;128(6):791-803. Epub 2014 Sep 28 PubMed.
View all comments by Neil CashmanMake a Comment
To make a comment you must login or register.