Cholesteryl Esters Hobble Proteasomes, Increase p-Tau
Quick Links
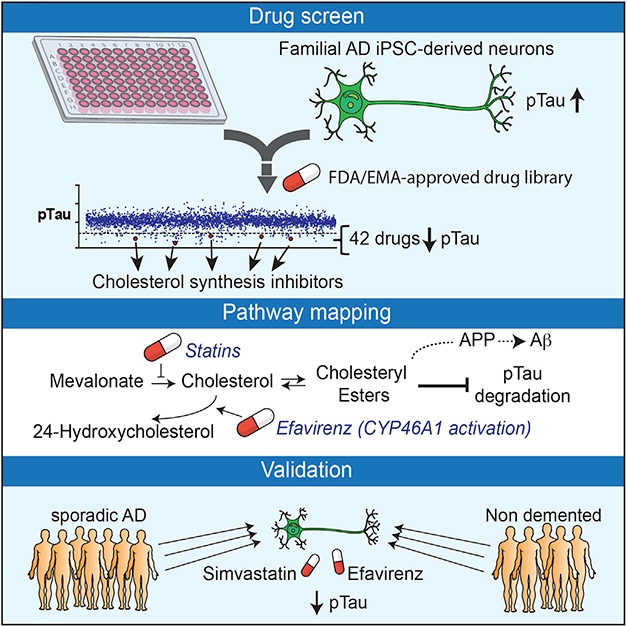
A cholesterol metabolite boosts accumulation of phosphorylated tau, according to a paper in the January 16 Cell Stem Cell. Researchers led by Lawrence Goldstein at the University of California, San Diego, reported that cholesteryl esters—which coalesce into lipid droplets for storage within the cell—boosted phosphorylated tau in neurons derived from people with Alzheimer’s disease. Reducing cholesteryl esters with efavirenz, a small-molecule drug that activates the enzyme 24-hydroxylase, reduced p-tau levels by ramping up its destruction by the proteasome.
- Cholesteryl esters boosted p-tau in human iPSC-derived neurons.
- Esters dampened proteasome activity.
- Efavirenz, which activates cholesterol hydroxylase, lowered p-tau.
Efavirenz is best known for inhibiting the reverse transcriptase of the HIV-1 virus, and is prescribed as an anti-AIDS medication. The drug has also been reported to reduce Aβ burden in animal models of amyloidosis. A Phase 1 clinical trial is underway to see if efavirenz safely boosts cholesterol hydroxylase activity in people with mild cognitive impairment. Goldstein and colleagues now propose that efavirenz could also lower p-tau in people with AD or other tauopathies, independent of its effect on APP.
“The study proposes that intraneuronal cholesterol dyshomeostasis could be an early trigger for both amyloidosis and tau pathological changes, albeit through distinct cascades of events,” commented Eloise Hudry of Massachusetts General Hospital in Boston.
Cholesteryl Esters Exacerbate p-tau, Aβ. Inhibitors of cholesterol synthesis lowered p-tau in neurons (top). Cholesteryl esters block p-tau degradation (middle). Statins and efavirenz lowered p-tau in human neurons by reducing ester concentrations (bottom). [Courtesy of van der Kant et al., 2019.]
The new findings emerged from an unbiased screen. Co-first authors Rik van der Kant and Vanessa Langness screened more than 1,600 FDA-approved or preclinical compounds for reduction of p-tau in neurons. Specifically, the researchers looked for changes in pThr231-tau/total tau ratios in induced pluripotent stem cell (iPSC)-derived neurons from a familial AD (FAD) patient carrying a duplication in the APP gene. Of 42 non-toxic hits that emerged, four were cholesterol-lowering statins: atorvastatin, simvastatin, fluvastatin, and rosuvastatin. Two lowered multiple isoforms of p-tau relative to total tau. The same happened in neurons from patients with late-onset sporadic AD and even in those from non-demented controls.
Statins squelch the first step in the cholesterol biosynthesis pathway by blocking the conversion of HMG-CoA into mevalonate. However, the researchers found that interfering with a much later step in the cholesterol pathway also reduced p-tau/tau ratio in the derived neurons. Specifically, the ratio dropped when the researchers dosed neurons with efavirenz, an allosteric activator of cholesterol 24-hydroxylase, aka CYP46A1. This enzyme, which is only expressed in the central nervous system, converts cholesterol into 24(S) hydroxycholesterol. Normally, neurons quickly export 24-OH-cholesterol, leaving less of it around to make cholesteryl esters. The neurons’ p-tau/tau ratio plummeted when the researchers overexpressed CYP46A1, or blocked Acyl-CoA cholesterol acyltransferase (ACAT), the enzyme that esterifies cholesterol, using the inhibitors avasimibe or K604. Conversely, the ratio shot up in response to exogenous cholesteryl esters. Together, these findings suggested that cholesteryl esters, not free cholesterol, elevated p-tau.
Why would this be? The scientists hypothesized that cholesteryl esters interfere with tau’s degradation. Though treatment with statins and efavirenz lowered the ratio of p-tau to tau, the absolute levels of both also dropped. However, when proteasome inhibitors were present, the drugs failed to lower tau. It’s not any type of degradation, though: Blockers of lysosomal/autophagosomal processing had no effect. Finally, the scientists found that statins or efavirenz increased levels of some proteasomal subunits, and revved up overall proteasome activity. Together, the data suggest that cholesterol esters obstruct the proteasome, letting tau build up. Goldstein does not know why this would change the p-tau/tau ratio.

Cholesteryl Esters Curb Proteasome. With a blocked proteasome, p-tau accumulates, but lowering cholesteryl esters with statins or efavirenz restored the organelle, reducing p-tau. [Courtesy of van der Kant et al., 2019.]
Goldstein and others emphasized that cholesteryl esters enhance the production of tau independently of Aβ and not downstream of it. The details of proteasome inhibition remain to be worked out, and Goldstein acknowledged that efavirenz and similar drugs might enhance the degradation of many other proteins, as well.
Separately, the researchers extended to FAD neurons a previous observation made in mouse models, namely, that statin treatment lowers secretion of Aβ (Oct 2004 news). However, the tau effect seems separate, because statins still reduced p-tau in isogenic APP-null neurons made by knocking out the gene in iPSCs derived from the patient harboring the APP duplication. The researchers also confirmed that cholesterol’s sway over Aβ secretion was mediated by its binding to a previously identified domain in the β-CTF portion of APP and influencing APP processing (Jun 2012 news). It is unclear whether cholesterol or its esters mediate this effect.
“This excellent study provides evidence that enhancing the activity of CYP46A1 and increasing the conversion of cholesteryl esters to 24-hydroxy-cholesterol may alleviate tau pathology,” wrote Per Svenningsson of the Karolinska Institute in Stockholm. The findings agree with Svenningsson’s recent study, which correlated 24-hydroxy-cholesterol levels with reduced tau in neuronal cells and in CSF in several tauopathies (Björkhem et al., 2018). “The concept of using efavirenz to treat tauopathies is novel and exciting,” he added.
Hudry thinks that activating cholesterol 24-hydroxylase, or downregulating ACAT, the enzyme that esterifies cholesterol, are potential therapeutic strategies. “Because CYP46A1 is only expressed in neuronal cells, it is an especially well-suited drug target, offering a unique opportunity to alleviate early AD neuropathological changes without interfering with the maintenance of cholesterol homeostasis in other cells or tissues,” she wrote (see comment below).
Irina Pikuleva of Case Western Reserve University in Cleveland is heading the mild cognitive impairment trial of efavirenz (see clinicaltrials.gov). It aims to find a dose that safely boosts 24-hydroxy-cholesterol levels in serum. Because this metabolite is only produced by CYP46A1 in the CNS, even traces of it in serum reflect activity of the enzyme in the brain, Pikuleva explained. Efavirenz is toxic when used to treat HIV, but Pikuleva and Goldstein believe much lower doses will suffice to reduce cholesteryl esters in the brain. In fact, at high doses, efavirenz inhibits, rather than activates, CYP46A1.
Pikuleva sees therapeutic potential for efavirenz in AD. She previously reported that the drug lowered Ab plaque burden and lessened memory loss in in 5xFAD mice and, on the flip side, other scientists reported that lowering CYP46A1 in the APP23 mouse brain worsened their plaque burden, tau phosphorylation, and memory loss (Mast et al., 2017; Djelti et al., 2015).—Jessica Shugart
References
News Citations
- ACAT and Mouse—Inhibiting Former Prevents AD-like Pathology in Latter
- Cholesterol Binds APP Fragment, May Direct It to Lipid Rafts
Research Models Citations
Paper Citations
- Björkhem I, Patra K, Boxer AL, Svenningsson P. 24S-Hydroxycholesterol Correlates With Tau and Is Increased in Cerebrospinal Fluid in Parkinson's Disease and Corticobasal Syndrome. Front Neurol. 2018;9:756. Epub 2018 Sep 7 PubMed.
- Mast N, Saadane A, Valencia-Olvera A, Constans J, Maxfield E, Arakawa H, Li Y, Landreth G, Pikuleva IA. Cholesterol-metabolizing enzyme cytochrome P450 46A1 as a pharmacologic target for Alzheimer's disease. Neuropharmacology. 2017 Sep 1;123:465-476. Epub 2017 Jun 24 PubMed.
- Djelti F, Braudeau J, Hudry E, Dhenain M, Varin J, Bièche I, Marquer C, Chali F, Ayciriex S, Auzeil N, Alves S, Langui D, Potier MC, Laprevote O, Vidaud M, Duyckaerts C, Miles R, Aubourg P, Cartier N. CYP46A1 inhibition, brain cholesterol accumulation and neurodegeneration pave the way for Alzheimer's disease. Brain. 2015 Aug;138(Pt 8):2383-98. Epub 2015 Jul 2 PubMed.
External Citations
Further Reading
Primary Papers
- van der Kant R, Langness VF, Herrera CM, Williams DA, Fong LK, Leestemaker Y, Steenvoorden E, Rynearson KD, Brouwers JF, Helms JB, Ovaa H, Giera M, Wagner SL, Bang AG, Goldstein LS. Cholesterol Metabolism Is a Druggable Axis that Independently Regulates Tau and Amyloid-β in iPSC-Derived Alzheimer's Disease Neurons. Cell Stem Cell. 2019 Mar 7;24(3):363-375.e9. Epub 2019 Jan 24 PubMed.
Annotate
To make an annotation you must Login or Register.
Comments
Karolinska Institutet
In this elegant work, van der Kant et al. performed a drug screen on hiPSC-derived AD neurons and identified, among others, four statins able to significantly decrease p-tau accumulation by reducing the levels of cholesteryl esters (CE). Interestingly, the same effect was mimicked by efavirenz, an allosteric activator of CYP46A1. The authors also found that while the effects of lowering CE on p-tau is mediated by the proteasome, the effect of CE on Aβ42 is mediated by the cholesterol-binding domain in APP.
This study highlights once again the importance of CYP46A1 and cholesterol turnover through the mevalonate pathway in the brain (Kotti et al., 2006). The findings are certainly in line with previous studies where activation of CYP46A1 in hippocampi of AD mice reduced Aβ plaques and restored spatial memory performances (Hudry et al., 2010), while inhibition of CYP46A1 in tau mice led to increased tau phosphorylation (Djelti et al., 2015). Treatment with low doses of efavirenz in AD mice also led to positive effects on Aβ reduction (Mast et al., 2017).
Altogether, targeting cholesterol metabolism may become a viable therapeutic approach for AD and other neurodegenerative diseases.
References:
Kotti TJ, Ramirez DM, Pfeiffer BE, Huber KM, Russell DW. Brain cholesterol turnover required for geranylgeraniol production and learning in mice. Proc Natl Acad Sci U S A. 2006 Mar 7;103(10):3869-74. PubMed.
Hudry E, Van Dam D, Kulik W, De Deyn PP, Stet FS, Ahouansou O, Benraiss A, Delacourte A, Bougnères P, Aubourg P, Cartier N. Adeno-associated virus gene therapy with cholesterol 24-hydroxylase reduces the amyloid pathology before or after the onset of amyloid plaques in mouse models of Alzheimer's disease. Mol Ther. 2010 Jan;18(1):44-53. PubMed.
Djelti F, Braudeau J, Hudry E, Dhenain M, Varin J, Bièche I, Marquer C, Chali F, Ayciriex S, Auzeil N, Alves S, Langui D, Potier MC, Laprevote O, Vidaud M, Duyckaerts C, Miles R, Aubourg P, Cartier N. CYP46A1 inhibition, brain cholesterol accumulation and neurodegeneration pave the way for Alzheimer's disease. Brain. 2015 Aug;138(Pt 8):2383-98. Epub 2015 Jul 2 PubMed.
Mast N, Lin JB, Anderson KW, Bjorkhem I, Pikuleva IA. Transcriptional and post-translational changes in the brain of mice deficient in cholesterol removal mediated by cytochrome P450 46A1 (CYP46A1). PLoS One. 2017;12(10):e0187168. Epub 2017 Oct 26 PubMed.
Harvard Medical School and Massachusetts General Hospital
Intraneuronal cholesterol dyshomeostasis at the crossroad between tau abnormal phosphorylation and amyloid-β secretion
This article by van der Kant and colleagues in Cell Stem Cell proposes that intraneuronal cholesterol dyshomeostasis could be an early trigger for both amyloidosis and tau pathological changes, albeit through distinct cascades of events. It is true that the link between cholesterol metabolism and Alzheimer’s disease (AD) has oftentimes been highlighted in epidemiological or experimental studies, but so far the exact underlying mechanisms have eluded investigations of human neuronal cells. Using an unbiased screen of over 1,600 FDA-approved and preclinical compounds in iPSC-derived neurons, the authors identified several statins as potent inhibitors of phospho-tau accumulation (across several tau phospho-epitopes and independent iPSC lines), an effect sustained after APP genetic ablation. Further investigation established that the accumulation of cholesteryl esters (CE, the storage form of cholesterol) rather than the content of intracellular free cholesterol, is at the center of a neuronal stress response that together triggers abnormal tau phosphorylation (via downregulation of the ubiquitin-proteasome system) and amyloid-β secretion (via APP interaction with cholesterol).
Those results suggest that either the downregulation of Acyl-CoA cholesterol acyltransferase (ACAT, the enzyme responsible for the conversion of cholesterol in CE) or the activation of cholesterol 24S-hydroxylase (CYP46A1, which metabolizes cholesterol into 24-hydroxycholesterol and allows its export out of the cell) in neurons could be therapeutically relevant to alleviate the early events of AD neuropathology. Because CYP46A1 is only expressed in neuronal cells, it is an especially well-suited drug target, offering a unique opportunity to alleviate early AD neuropathological changes without interfering with the maintenance of cholesterol homeostasis in other cells or tissues.
Power of the study
The idea of modulating cholesterol metabolism to inhibit Aβ or tau neurotoxicity is not novel and has been previously explored in mouse models of amyloidosis and tauopathy, either via inhibiting ACAT activity (Hutter-Paier et al., 2004; Shibuya et al., 2015) or by overexpressing CYP46A1 in neurons (Hudry et al., 2010; Burlot et al., 2015). Nonetheless, the present study stands out for several reasons: 1) the authors identified cholesterol metabolism and CE intraneuronal accumulation as a central “druggable axis” for early signs of AD after an unbiased screen of over 1,600 compounds (a non-hypothesis driven strategy); 2) the reported effects of statins to decrease the levels of several tau phospho-epitopes were validated in familial AD- (APP duplication), sporadic AD patient- and non-demented control neurons (therefore suggesting that this approach may be impactful in all AD cases); 3) this protective impact persists in absence of APP expression (highlighting independent pathways through which CE triggers amyloid and tau pathological changes, without the necessity for a cause/consequence relationship between those neurotoxic species); 4) this study reveals how cholesterol dyshomeostasis could drive pathology in a cell-autonomous manner in human neurons, independently of the presence of astrocytes (well-known “suppliers” of cholesterol to mature neurons in the brain).
Perspective of the study
This work should be put in perspective with the many epidemiological studies that have tried to address the therapeutic potential of statins on dementia and AD cognitive decline (recently reviewed in Mejias-Trueba et al., 2018; Daneschvar et al., 2015). The results of those studies have been somewhat inconsistent, and no clear consensus has been established so far. Those discrepancies could be explained by different blood-brain barrier permeabilities (able to cross or not the blood-brain barrier), the stage at which they were administered, or the distinct pharmacological effects each of those molecules can trigger throughout the body.
In any case, the present findings by van der Kant and colleagues warrant a deeper investigation of the complex interactions between cholesterol dyshomeostasis and AD neurodegeneration, shedding new light on the significant cell-autonomous impact of CE metabolism in neurons. Whether or not Apolipoprotein E status may further modulate this phenotype is an intriguing hypothesis to explore, especially considering that the latest publication by Dr. Tsai’s lab (MIT) demonstrating dramatic differences in the transcriptional profiles of APOE4- or APOE3-iPSC-derived neurons (Lin et al., 2018).
Finally, the authors did not comment on the other hits that had emerged from their screening effort to select Thr231-phospho-tau reducing agents (42 were initially identified, including three microtubule-interacting compounds and four statins), and one may wonder if other important molecular cascades involved in the reduction of pathological post-translational tau modifications could emerge next.
References:
Hutter-Paier B, Huttunen HJ, Puglielli L, Eckman CB, Kim DY, Hofmeister A, Moir RD, Domnitz SB, Frosch MP, Windisch M, Kovacs DM. The ACAT inhibitor CP-113,818 markedly reduces amyloid pathology in a mouse model of Alzheimer's disease. Neuron. 2004 Oct 14;44(2):227-38. PubMed.
Shibuya Y, Niu Z, Bryleva EY, Harris BT, Murphy SR, Kheirollah A, Bowen ZD, Chang CC, Chang TY. Acyl-coenzyme A:cholesterol acyltransferase 1 blockage enhances autophagy in the neurons of triple transgenic Alzheimer's disease mouse and reduces human P301L-tau content at the presymptomatic stage. Neurobiol Aging. 2015 Jul;36(7):2248-59. Epub 2015 Apr 7 PubMed.
Hudry E, Van Dam D, Kulik W, De Deyn PP, Stet FS, Ahouansou O, Benraiss A, Delacourte A, Bougnères P, Aubourg P, Cartier N. Adeno-associated virus gene therapy with cholesterol 24-hydroxylase reduces the amyloid pathology before or after the onset of amyloid plaques in mouse models of Alzheimer's disease. Mol Ther. 2010 Jan;18(1):44-53. PubMed.
Burlot MA, Braudeau J, Michaelsen-Preusse K, Potier B, Ayciriex S, Varin J, Gautier B, Djelti F, Audrain M, Dauphinot L, Fernandez-Gomez FJ, Caillierez R, Laprévote O, Bièche I, Auzeil N, Potier MC, Dutar P, Korte M, Buée L, Blum D, Cartier N. Cholesterol 24-hydroxylase defect is implicated in memory impairments associated with Alzheimer-like Tau pathology. Hum Mol Genet. 2015 Nov 1;24(21):5965-76. Epub 2015 Sep 10 PubMed.
Mejías-Trueba M, Pérez-Moreno MA, Fernández-Arche MÁ. Systematic review of the efficacy of statins for the treatment of Alzheimer's disease. Clin Med (Lond). 2018 Feb;18(1):54-61. PubMed.
Daneschvar HL, Aronson MD, Smetana GW. Do statins prevent Alzheimer's disease? A narrative review. Eur J Intern Med. 2015 Nov;26(9):666-9. Epub 2015 Sep 2 PubMed.
Lin YT, Seo J, Gao F, Feldman HM, Wen HL, Penney J, Cam HP, Gjoneska E, Raja WK, Cheng J, Rueda R, Kritskiy O, Abdurrob F, Peng Z, Milo B, Yu CJ, Elmsaouri S, Dey D, Ko T, Yankner BA, Tsai LH. APOE4 Causes Widespread Molecular and Cellular Alterations Associated with Alzheimer's Disease Phenotypes in Human iPSC-Derived Brain Cell Types. Neuron. 2018 Jun 27;98(6):1141-1154.e7. Epub 2018 May 31 PubMed.
Brigham and Women’s Hospital, Harvard Medical School
Dartmouth Medical School
We read this paper with great interest. This work provides a novel link between cholesterol esters (CE) with tau pathology in Alzheimer's disease patient-derived neurons. According to the authors, the CE effects on tau proteostasis are independent of APP and Aβ. The authors conclude that decreasing CE and/or increasing 24(S)-hydroxycholesterol is a viable strategy to reduce accumulation of pTau and Aβ in AD patients. We fully concur with this notion.
Interestingly, the working model described in this work shows a shared pathway between cholesterol to be esterified by the esterification enzyme ACAT, and cholesterol to be converted to 24(S)-hydroxycholesterol by the enzyme Cyp46A1. In 2010, we reported that ACAT1 gene ablation increases 24(S)-hydroxycholesterol content in the brain and ameliorates amyloid pathology in mice with AD, suggesting that ACAT1 and Cyp46a1 share the same cholesterol substrate pool in the AD mouse brain. We are pleased that the same concept is applicable in the AD patient iPSC derived human neuronal cells.
Cathy Chang at the Geisel School of Medicine at Dartmouth also contributed to this comment.
References:
Bryleva EY, Rogers MA, Chang CC, Buen F, Harris BT, Rousselet E, Seidah NG, Oddo S, Laferla FM, Spencer TA, Hickey WF, Chang TY. ACAT1 gene ablation increases 24(S)-hydroxycholesterol content in the brain and ameliorates amyloid pathology in mice with AD. Proc Natl Acad Sci U S A. 2010 Feb 16;107(7):3081-6. PubMed.
Make a Comment
To make a comment you must login or register.