Can Anna Karenina Help Scientists Find Proteins That Cause Alzheimer’s?
Quick Links
Many genes and proteins have been linked to Alzheimer’s, but which ones directly contribute to disease? This question has been hard to answer. In the December 11 Cell Genomics, scientists led by Zhonghua Liu at Columbia University in New York City suggested a new way to approach it. Mendelian randomization post-selection inference looks for different genetic variants that similarly affect the amount of a given protein and the risk for disease. Evoking an analogy to the “Anna Karenina” principle, MR-SPI posits that if different protein quantitative trait loci all correlate equally strongly with AD, then the protein in question does, in fact, contribute to Alzheimer’s. Named after Leo Tolstoy’s novel’s famous opening statement that “All happy families are alike; each unhappy family is unhappy in its own way,” the principle in this instance infers that all true causal variants will have similar effects, while false positives will not.
- Multiple AD genetic variants can affect a single protein in the same way.
- That similarity should reflect underlying biology, suggesting a direct role in disease.
- This principle led to seven AD proteins; one of them new.
In a proof-of-concept for this method, the authors identified 52 pQTLs that modulate levels of seven proteins predicted to be causal (image below). Six are encoded by known AD genes, including TREM2 and CD33, while the growth factor receptor RET is new. The authors believe MR-SPI will be useful for identifying potential biomarkers or therapeutic targets. Further, identifying causal variants can also help decipher how they promote disease, for example by altering a protein’s shape. The authors predicted three of these proteins would have a changed structure, based on causal missense variants in the coding region.
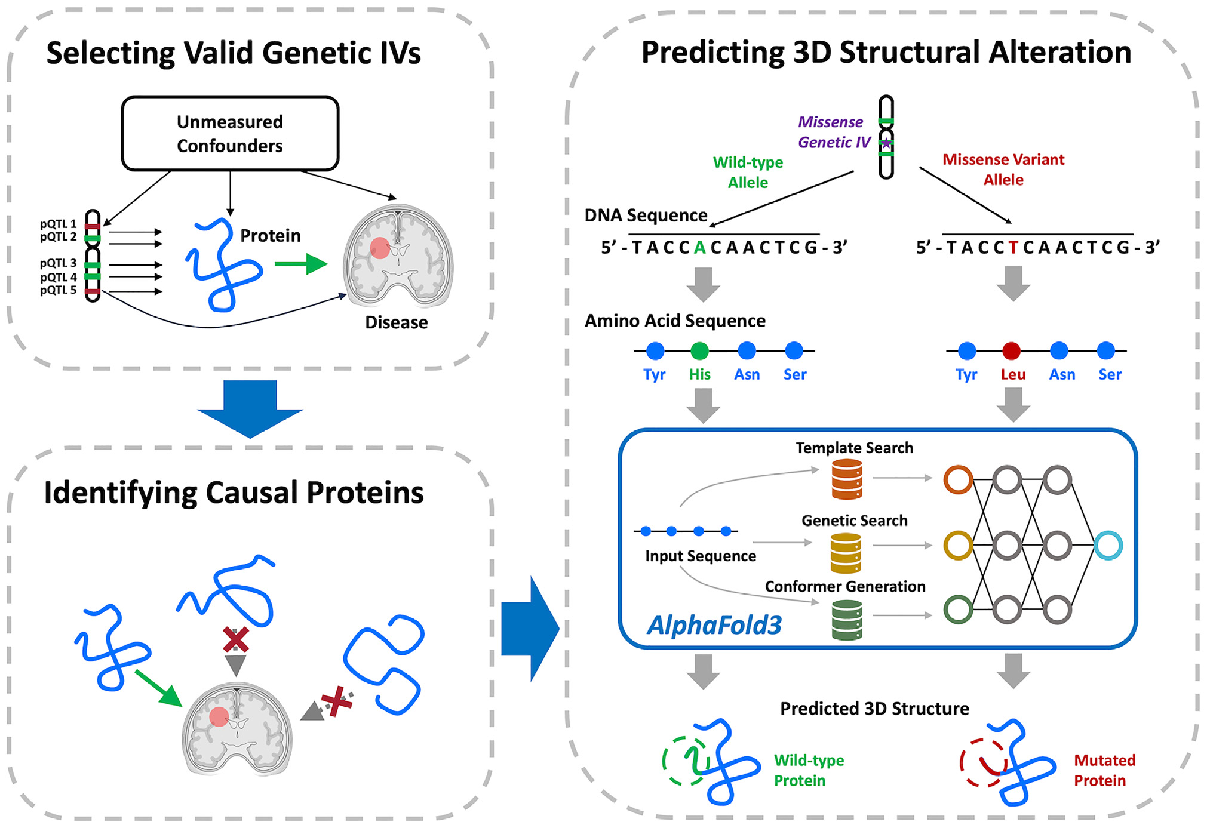
Pegging Causes. A proposed pipeline first identifies pQTLs related to disease (upper left), then winnows out those unlikely to be causal (lower left), and finally predicts how missense pQTLs affect protein structure (right). [Courtesy of Yao et al., Cell Genomics.]
Erik Johnson at Emory University, Atlanta, agreed MR-SPI is an advance. “The authors’ algorithm addresses a number of challenges with Mendelian randomization using proteomics data. An advantage of their approach is the robust way pQTLs are selected as instrumental variables,” he wrote to Alzforum (comment below).
Mendelian randomization uses genetic variants to infer that a protein causes disease. The reasoning goes that if an SNP associates with the amount of a protein, and with the likelihood of getting a certain disease, then that protein probably plays a direct role in pathology (image below). However, confounding factors can foil this method, for example when pQTLs affect multiple proteins.

Group Process. When multiple genetic variants, e.g. pQTLs 1, 2, and 3 (left) similarly affect protein level (x axis) and disease (y axis), they likely reflect real biology. If pQTLs cluster poorly, e.g., 4 and 5 (right), they are probably not related to the disease. [Courtesy of Yao et al., Cell Genomics.]
To find true causal variants, first author Minhao Yao at the University of Hong Kong applied the Anna Karenina principle to U.K. Biobank proteomic and genetic data from 54,306 participants. The dataset comprised 912 plasma proteins, and so could have missed AD proteins that are not abundant in blood. This analysis turned up 10,248 total pQTLs; curiously, only 10 percent of these variants acted on a nearby gene, with the rest acting on distant ones. Next, the authors correlated these pQTLs with AD GWAS data from the International Genomics of Alzheimer’s Project study, to find SNPs linked both to protein level and disease (Mar 2019 news). Then they winnowed these SNPs down to the likely causal variants using the Anna Karenina method.
They identified seven proteins with a causal link to AD. Six of them—TREM2, CD33, CD55, EPHA1, PILRA, and PILRB—were known AD risk factors. The last, receptor tyrosine kinase (RET), has not been previously linked to Alzheimer’s. This protein is best known as an oncogene involved in cell survival. RET is present in mitochondria, and so may be involved in age-related declines in energy metabolism.

Small Change, Big Effect. An SNP that changes an A to a G in the coding region of CD33 substitutes a glycine (yellow) for an arginine (green), altering the protein from its original shape (blue) to a new one (red). [Courtesy of Yao et al., Cell Genomics.]
Knowing the causal variants can offer clues to disease mechanisms, such as whether too much or too little of the protein leads to disease, the authors noted. Some causal variants directly affect protein structure. Three of the proteins, TREM2, CD33, and PILRA, had missense pQTLs in their coding regions. The authors used the software package AlphaFold3, developed by Google DeepMind and Isomorphic Labs, to predict how these variants would affect protein folding (Jumper et al., 2021). For example, CD33 changes shape due to an SNP that substitutes G for A, resulting in incorporation of a glycine instead of an arginine (image at left).
The authors believe their method could be applied to dissect the causes of many other complex diseases as well.—Madolyn Bowman Rogers
References
Paper Citations
- Jumper J, Evans R, Pritzel A, Green T, Figurnov M, Ronneberger O, Tunyasuvunakool K, Bates R, Žídek A, Potapenko A, Bridgland A, Meyer C, Kohl SA, Ballard AJ, Cowie A, Romera-Paredes B, Nikolov S, Jain R, Adler J, Back T, Petersen S, Reiman D, Clancy E, Zielinski M, Steinegger M, Pacholska M, Berghammer T, Bodenstein S, Silver D, Vinyals O, Senior AW, Kavukcuoglu K, Kohli P, Hassabis D. Highly accurate protein structure prediction with AlphaFold. Nature. 2021 Aug;596(7873):583-589. Epub 2021 Jul 15 PubMed.
Further Reading
Primary Papers
- Yao M, Miller GW, Vardarajan BN, Baccarelli AA, Guo Z, Liu Z. Deciphering proteins in Alzheimer's disease: A new Mendelian randomization method integrated with AlphaFold3 for 3D structure prediction. Cell Genom. 2024 Dec 11;4(12):100700. Epub 2024 Dec 4 PubMed.
Annotate
To make an annotation you must Login or Register.
Comments
Emory University
Yao et al. propose an interesting Mendelian randomization (MR) method that incorporates a robust, instrumental-variable selection procedure and downstream integration with AlphaFold to predict the effects of missense variants on 3D structure. The authors’ algorithm addresses a number of challenges using MR with proteomics data. An advantage of their approach is the robust way protein quantitative trait loci (pQTLs) are selected as instrumental variables in MR. One issue to consider for this selection is genetic variation, which could lead to an apparent change in exposure/protein level, simply due to loss, or gain, of binding affinity of the reagent to the protein; this is less of a problem with direct methods, such as mass spectrometry. The authors do not comment on this issue in the article, but it would be interesting to see how their algorithm handles such instances.…More
A nice feature of the algorithm is incorporation of three-dimensional structure prediction with AlphaFold to see how missense variants may ultimately affect protein structure. Incorporation of co-localization analysis into the package, as the authors plan to do, will provide even more utility. The MR-SPI method represents an advance for proteomics-based MR. Congratulations to the authors.
University of Eastern Finland
Yao et al. have developed and tested a novel Mendelian randomization method that is integrated to the three-dimensional protein structure prediction tool. Specifically, the authors first established an MR method called MR-SPI that selects valid protein quantitative loci (pQTL). It is based on the principal of Leo Tolstoy’s dictum “all happy families are alike; each unhappy family is unhappy in its own way” to identify causal protein biomarkers for health outcomes. This MR framework selects valid pQTLs as instrumental variables to be subsequently used also for the three-dimensional structural protein analysis (AlphaFold3). After testing the performance of MR-SPI using simulated data alongside other established MR methods, the authors applied this all-in-one pipeline to discover potential causal plasma protein biomarkers associated with Alzheimer’s disease (AD). For that purpose, plasma-based proteomics data and ~23 million imputed autosomal variants across ~1,500 proteins from the U.K. Biobank were retrieved from a cohort comprising ~54,000 participants. After filtering steps to select independent and strongly associated pQTL instrument variables, the authors used summary statistics for clinically diagnosed AD and AD by proxy from a meta-analysis of GWAS’s originating from the Jansen et al. study (2019). As a result, MR-SPI identified seven plasma proteins (CD33, CD55, EPHA1, PILRA, PILRB, RET, and TREM2) that either were positively or negatively associated with the risk of AD. Furthermore, the following AlphaFold3 and gene ontology enrichment analyses revealed specific local protein changes as well as common biological readouts linked to identified plasma targets.…More
Collectively, the stepwise process of how the MPI-SPI framework selects, and validates by voting, the most relevant instrument variables to the final estimation of causal effects is extremely insightful. This provides an alternative strategy to identify potential AD-associated biomarkers.
Given the rapidly accumulating genetic and omics data in different databases and biobanks world-wide, it is of utmost importance to leverage these kinds of integrated MR pipelines to pinpoint disease-related proteins to be further applied for biomarker development. Although there are also some limitations noted by the authors, the MR-SPI pipeline indeed holds great potential not only for detecting causal protein biomarkers associated with diseases, but also for the characterization of underlying disease mechanisms.
References:
Jansen IE, Savage JE, Watanabe K, Bryois J, Williams DM, Steinberg S, Sealock J, Karlsson IK, Hägg S, Athanasiu L, Voyle N, Proitsi P, Witoelar A, Stringer S, Aarsland D, Almdahl IS, Andersen F, Bergh S, Bettella F, Bjornsson S, Brækhus A, Bråthen G, de Leeuw C, Desikan RS, Djurovic S, Dumitrescu L, Fladby T, Hohman TJ, Jonsson PV, Kiddle SJ, Rongve A, Saltvedt I, Sando SB, Selbæk G, Shoai M, Skene NG, Snaedal J, Stordal E, Ulstein ID, Wang Y, White LR, Hardy J, Hjerling-Leffler J, Sullivan PF, van der Flier WM, Dobson R, Davis LK, Stefansson H, Stefansson K, Pedersen NL, Ripke S, Andreassen OA, Posthuma D. Genome-wide meta-analysis identifies new loci and functional pathways influencing Alzheimer's disease risk. Nat Genet. 2019 Mar;51(3):404-413. Epub 2019 Jan 7 PubMed.
Make a Comment
To make a comment you must login or register.