Aβ Dimers Block Glutamate Uptake, Fire Up Synapses
Quick Links
Soluble Aβ sends neurons into a hyperactive frenzy, but scientists don’t know why. New evidence suggests that the peptide causes the excitatory neurotransmitter glutamate to build up in synapses, making neurons hyperactive. In the August 9 Science, researchers led by Arthur Konnerth, Technische Universität München, Germany, suggest that after a neuron fires, Aβ dimers block clearance of glutamate released into the synaptic cleft. This makes the neurons even more active. The evidence comes from both live mice and hippocampal slices. The results may explain overactive neurons seen in mouse models of amyloidosis and in people with prodromal AD.
- Aβ dimers, both synthetic and human-derived, make neurons hyperexcitable.
- Compounds that block glutamate reuptake cause similar hyperexcitability.
- Aβ dimers may interfere with glutamate clearance, leaving more of the neurotransmitter in the synapse.
“This is a nice paper that mostly reinforces what we know about the interaction of Aβ at the synapse, but adds significant insight into the interaction of low-molecular-weight soluble oligomers of amyloid with specific components of the synapse,” wrote Brian Bacskai, Massachusetts General Hospital, Boston.
Scientists have known for years that various forms of soluble Aβ make neurons more excitable (May 2012 news). But the toxic species of Aβ and the mechanism by which they cause the hyperactivity are still in question. One hypothesis, from the lab of co-author Dominic Walsh at Brigham and Women’s Hospital, Boston, contends that Aβ dimers are the most toxic species (Nov 2018 conference news). As for the mechanism, evidence suggests glutamate might be involved. Researchers from Dennis Selkoe’s lab at the Brigham reported that Aβ interfered with reuptake of glutamate from the synapse, and that this impaired plasticity in hippocampal slices (Li et al., 2009; Li et al., 2011). Might poor reuptake cause hyperexcitability as well, and does this take place in live mice?
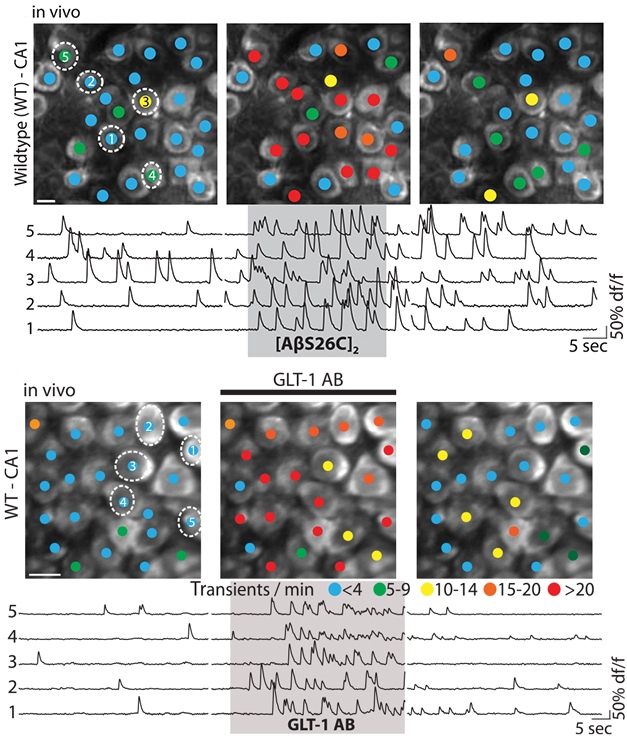
Exciting Compounds. Calcium imaging shows that active neurons (blue and green dots at left) fire more often (red, orange, yellow; middle) with the addition of either synthetic Aβ dimers ([AβS26C]2) or an antibody that impairs glutamate reuptake (GLT-1 AB). After washout, cell activity returns to normal (right panels). [Imaging courtesy of AAAS/Science.]
First author Benedikt Zott and colleagues started to address this by testing how neurons in 2-month-old wild-type mice responded to injected Aβ dimers. They used a special cranial window to monitor neurons by two-photon Ca2+ imaging while they pipetted various substances onto the hippocampal surface (Busche et al., 2012). Synthetic Aβ dimers quadrupled Ca2+ transients relative to baseline (see image above).
Interestingly, the same dimers applied to hippocampal slices had no effect. However, if the researchers stimulated the hippocampal slices by adding glutamate, blocking inhibitory inputs, or elevating the extracellular K+ concentration, Aβ dimers revved up the activity even further, just as they did it vivo. The results suggested that the mechanism whereby Aβ drives hyperexcitability relies on synapses that are already active.
To see if impaired reuptake of glutamate might be responsible for the Aβ effect, the authors tried to mimic it by pipetting the glutamate uptake blocker DL-threo-b-benzyloxyaspartic acid (TBOA) onto the hippocampal CA1 region of wild-type mice. They saw a similar quadrupling of Ca2+ transients as in Aβ-treated neurons. Further, both the Aβ dimers and TBOA transiently increased the extracellular glutamate concentration, suggesting they acted by a similar mechanism.
Still, TBOA might act via a different mechanism. To test this, Zott repeated the TBOA experiment in 2-month-old APP23xPS45 mice (Busche et al., 2008). These animals are too young to have developed plaques but old enough to have plenty of soluble Aβ in their brains and their hippocampi are markedly hyperactive. TBOA had no effect on hippocampal activity. The authors believe this is because the endogenous Aβ had already fully blocked glutamate reuptake.
How is that block achieved? Because astrocytes normally clear glutamate from synapses, the researchers wondered if Aβ might affect GLT-1, the main excitatory amino-acid transporter in these cells. GLT1 hangs out on the plasma membrane of astroglial projections that protrude into synapses. When a neuron fires, GLT1 quickly binds the newly released glutamate, then immediately diffuses along the projections away from the synapse, dragging glutamate with it. The neurotransmitter then passes through the transporter into the astrocyte (Murphy-Royal et al., 2015). Researchers can impair this process using an antibody to cross-linking GLT-1, anchoring the transporters in the membrane. Treating the CA1 region of hippocampi in wild-type mice with this antibody induced hyperexcitability on par with Aβ dimers (see image above), again suggesting to the authors that the two work via a similar mechanism.
The authors repeated their experiments using Aβ dimers isolated from human brains. These made mouse neurons hyperactive at lower concentrations than did synthetic ones, suggesting the human dimers are more potent.
Together, the results suggest that Aβ dimers work in a vicious cycle to make active neurons even more active by interfering with glutamate uptake. However, Keith Vossel, University of Minnesota, Minneapolis, pointed out that the paper relied on circumstantial evidence. Direct proof that Aβ obstructs the diffusion of GLT-1 was lacking. Demonstrating that hippocampal slices need to have baseline activity before they are susceptible to Aβ-induced hyperexcitability, was important, he said.
Selkoe thought it promising that Aβ could interfere with diffusion of glutamate receptors. "A key next step will be to decipher at the biochemical level precisely how the oligomers block glutamate entry into local astrocytes and neurons,” he wrote in an accompanying editorial. He thinks that glutamine antagonists, or agents such as ceftriaxone, which boosts the number and function of glutamine transporters, could be clinically useful if given at the earliest stages of Alzheimer’s. Combined with therapies that reduce Aβ-oligomers, such as secretase inhibitors, they might work together to lower hyperactivity at the very earliest stages of the disease. The paper “highlights that efforts to identify and test anti-Aβ agents need to be actively pursued, even as attention appropriately turns to non–amyloid-based AD interventions,” he wrote.—Gwyneth Dickey Zakaib
References
News Citations
- Soluble Aβ Takes Blame for Hyperactive Neurons in Mouse Brain
- Toxic Stew of Aβ Dimers Hides Out in Human Plaques
Paper Citations
- Li S, Hong S, Shepardson NE, Walsh DM, Shankar GM, Selkoe D. Soluble oligomers of amyloid Beta protein facilitate hippocampal long-term depression by disrupting neuronal glutamate uptake. Neuron. 2009 Jun 25;62(6):788-801. PubMed.
- Li S, Jin M, Koeglsperger T, Shepardson NE, Shankar GM, Selkoe DJ. Soluble Aβ oligomers inhibit long-term potentiation through a mechanism involving excessive activation of extrasynaptic NR2B-containing NMDA receptors. J Neurosci. 2011 May 4;31(18):6627-38. PubMed.
- Busche MA, Chen X, Henning HA, Reichwald J, Staufenbiel M, Sakmann B, Konnerth A. Critical role of soluble amyloid-β for early hippocampal hyperactivity in a mouse model of Alzheimer's disease. Proc Natl Acad Sci U S A. 2012 May 29;109(22):8740-5. Epub 2012 May 16 PubMed.
- Busche MA, Eichhoff G, Adelsberger H, Abramowski D, Wiederhold KH, Haass C, Staufenbiel M, Konnerth A, Garaschuk O. Clusters of hyperactive neurons near amyloid plaques in a mouse model of Alzheimer's disease. Science. 2008 Sep 19;321(5896):1686-9. PubMed.
- Murphy-Royal C, Dupuis JP, Varela JA, Panatier A, Pinson B, Baufreton J, Groc L, Oliet SH. Surface diffusion of astrocytic glutamate transporters shapes synaptic transmission. Nat Neurosci. 2015 Feb;18(2):219-26. Epub 2015 Jan 12 PubMed.
Further Reading
Primary Papers
- Zott B, Simon MM, Hong W, Unger F, Chen-Engerer HJ, Frosch MP, Sakmann B, Walsh DM, Konnerth A. A vicious cycle of β amyloid-dependent neuronal hyperactivation. Science. 2019 Aug 9;365(6453):559-565. PubMed.
- Selkoe DJ. Early network dysfunction in Alzheimer's disease. Science. 2019 Aug 9;365(6453):540-541. PubMed.
Annotate
To make an annotation you must Login or Register.
Comments
BrightFocus Foundation
This paper describes impressive experimentation with convincing results, though I would be interested in seeing more discussion of chronic glutamate toxicity in AD. Before investing further in therapeutic strategies targeting reduction of Aβ-oligomers, there must be evaluation of compensatory processes for gradual increases in hyperexcitability in aging. This is beyond the scope of this work, but relevant to the overall translatability of the research. Neurotoxicity of glutamargeric hyperexcitation is typically an acute event, though there is an existing body of research for chronic cases (reviewed by Lewerenz & Maher, 2015).
Alzheimer's & Dementia
This new story is noteworthy not only for the findings but also for the lack of adequate exposition [or the significance] of the downstream sequels to a neuron ‘driven’ into an “…hyperactive frenzy…”. One of the longstanding challenges for all the major theories on putative mechanisms of neurodegeneration or dementia or Alzheimer’s disease [AD], including the ‘amyloid’ hypothesis, has been the lack of compelling explanation on how the particular hypothesis influences the functioning/performance of a neuron; as a constituent of a network or a system. The significance of this question stems from the premise that progressive decline in the performance/functioning of various neural networks is the most proximal and crucial event that accounts for the clinical features of chronic brain disorders associated with neurodegeneration; including dementia and AD. Although various theories on the origins of these conditions usually start with differing assumptions, virtually all models of pathogenesis invoke some form of deficit in the functional connectivity of neural networks. However, the precise molecular mechanism for linking the putative biological mechanism of the ‘disease’ with its ‘clinical’ expression or special features of the condition has remained an ill-defined ‘black box’. Recent studies from groups such as those headed by Cindy Lemere and Dennis Selkoe groups’ [see- Hong S, Beja-Glasser VF, Nfonoyim BM, Frouin A, Li S, Ramakrishnan S, Merry KM, Shi Q, Rosenthal A, Barres BA, Lemere CA, Selkoe DJ, Stevens B. Complement and microglia mediate early synapse loss in Alzheimer mouse models. Science. 2016 Mar 31] have begun to shed light on this ‘black box’ by providing explanations for plausible mechanistic links between upstream events and neuronal function by proposing the possibility of a compliment/microglia mediated neuronal dysfunction. In this context, the finding by Zott et al that soluble Aβ dimers induce hyperexcitability in neurons, similar to those caused by blocking glutamate reuptake, is very significant. This study provides yet another important insight about the workings of the ‘Black Box.’ In light of the sequential failure of therapy development paradigms [e.g., clinical trials] or, based on the ‘amyloid hypothesis,’ the urgency of uncovering and considering alternative plausible mechanisms that mediate neuronal function-dysfunction has become more vital than ever. Until recently, alternative ideas on the origins of dementia-AD and/or potential targets for therapy development were largely overshadowed by the amyloid hypothesis. But, now with the changing climate for drug discovery-development, along with the search for better or more precise explanations of molecular mechanism that mediate neuronal functioning, hitherto overlooked ideas or theories may get more attention and flourish. The Calcium Hypothesis [1, 2] is a good case-study to illustrate this point. The relationship between ‘amyloid’ and calcium dyshomeostasis has been known for some time [see Arispe N, Pollard HB, Rojas E. The ability of amyloid b-protein [AbP (1-40)] to form Ca2+ channels provides a mechanism for neuronal death in Alzheimer’s disease. Ann N Y Acad Sci 1994; 747:256–66]. Now, Zott et al provide further evidence that the so called ‘amyloid toxicity’ is actually mediated by calcium dysregulation. Perhaps, this will generate greater interest in the ‘Calcium Hypothesis and the proposition that cytosol calcium dyshomeostasis is represents the final common path for neuronal dysfunction [i.e., continued pruning of dendritic arbors, loss of synapses, and decrements in repair and restoration]. Thus, provides the most proximal mechanism [and most plausible explanation] for the expression of clinical features of l neurodegenerative disorders such as dementia and AD [3]. References: 1. Khachaturian ZS. Calcium hypothesis of Alzheimer’s disease and brain aging. In: Calcium hypothesis of aging and dementia. Disterhoft JF, Willem HG, Treber J, Khachaturian ZS. Eds. Ann N Y Acad Sci 1994;747:1–11. 2. Alzheimer’s Association Calcium Hypothesis Workgroup. Calcium Hypothesis of Alzheimer’s disease and brain aging: A framework for integrating new evidence into a comprehensive theory of pathogenesis’. Alzheimers Dement 2017;13:178–82. 3. Khachaturian ZS, Kuller LH, Khachaturian AK. Editorial: Strategic goals and roadmap for dementia prevention by stroke Prevention. Alzheimers Dementia 2019;15: 865-869.…More
University of Kentucky
This paper suggests that defects in GLT-1 (also known as EAAT2) in astrocytes lead to accumulation of synaptic glutamate, which causes hyperactivity, leading to excitotoxicity of and Ca2+ entry to neurons. Nearly two decades ago, our laboratory demonstrated that, in AD brain, GLT-1 was oxidatively modified by the lipid peroxidation product, 4-hydroxynonenal (HNE) to more than 70% more than that in control brain (Lauderback et al., 2001). Moreover, this paper showed that addition of Abeta1-42 to synaptic preparations containing pre- and post-synaptic membranes reproduced the same finding as in AD brain. Our prior studies had shown that HNE covalent modification of Cys, His, or Lys residues of synaptic proteins following Michael addition reactions alters their conformation (Subramaniam et al.,1997). This, essentially, always led to decreased function (Butterfield and Halliwell, 2019). Masliah and colleagues showed that GLT-1 activity was decreased in AD brain (Masliah et al., 1996). Therefore, we hypothesize that the elegant studies led by Prof. Konnerth have a basis derived from HNE-oxidative modification of GLT-1 structure and function. …More
References:
Lauderback CM, Hackett JM, Huang FF, Keller JN, Szweda LI, Markesbery WR, Butterfield DA. The glial glutamate transporter, GLT-1, is oxidatively modified by 4-hydroxy-2-nonenal in the Alzheimer's disease brain: the role of Abeta1-42. J Neurochem. 2001 Jul;78(2):413-6. PubMed.
Subramaniam R, Roediger F, Jordan B, Mattson MP, Keller JN, Waeg G, Butterfield DA. The lipid peroxidation product, 4-hydroxy-2-trans-nonenal, alters the conformation of cortical synaptosomal membrane proteins. J Neurochem. 1997 Sep;69(3):1161-9. PubMed.
Butterfield DA, Halliwell B. Oxidative stress, dysfunctional glucose metabolism and Alzheimer disease. Nat Rev Neurosci. 2019 Mar;20(3):148-160. PubMed.
Masliah E, Alford M, DeTeresa R, Mallory M, Hansen L. Deficient glutamate transport is associated with neurodegeneration in Alzheimer's disease. Ann Neurol. 1996 Nov;40(5):759-66. PubMed.
Make a Comment
To make a comment you must login or register.